Мягкие капсулы, содержащие палоносетрона гидрохлорид, имеющие улучшенную стабильность и биологическую доступность
Номер патента: 16455
Опубликовано: 30.05.2012
Авторы: Кальдерари Джорджо, Бонадео Даниеле, Бралья Энрико, Бралья Риккардо













Формула / Реферат
1. Мягкая желатиновая капсула для перорального введения, содержащая:
a) мягкую желатиновую наружную оболочку, имеющую проницаемость для кислорода, меньшую, чем примерно 1,0´10-3 мл×см/(см2×24 ч×атм); и
b) липофильную жидкую композицию внутренней начинки, содержащую:
i) больше чем примерно 50 мас.% одного или нескольких липофильных компонентов;
(ii) от примерно 1 до примерно 20 мас.% воды, перемешанной или гомогенизированной в указанных одном или нескольких липофильных компонентах;
(iii) от примерно 0,05 до примерно 2,0 мг палоносетрона гидрохлорида, солюбилизированного или диспергированного в указанной воде; и
iv) поверхностно-активное вещество,
где указанная капсула при пероральном приеме в голодном состоянии демонстрирует фармакокинетику, которая биологически эквивалентна препарату, имеющему более чем 95% абсолютную биологическую доступность, причем биологическая эквивалентность устанавливается с помощью 90% доверительного интервала для AUC, который находится в пределах между 80 и 125%.
2. Капсула по п.1, в которой указанная композиция внутренней начинки содержит:
a) от 0,5 до 1,0 мг палоносетрона гидрохлорида и
b) солюбилизирующее эффективное количество жидкости, содержащей липофильный наполнитель и воду.
3. Мягкая желатиновая капсула по п.1, содержащая глицерин в указанной наружной оболочке и указанную композицию внутренней начинки.
4. Мягкая желатиновая капсула по п.1, в которой:
a) указанная композиция внутренней начинки дополнительно содержит антиоксидант или восстанавливающий агент;
b) указанный палоносетрон содержит меньше чем примерно 1 мас.% (3S)-3-[(3aS)-1-оксо-2,3,3а,4,5,6-гексагидро-1H-бензо[de]изохинолин-2-ил]-1-азониабицикло[2.2.2]октан-1-олат.
5. Капсула по п.1, демонстрирующая фармакокинетику при пероральном приеме в голодном состоянии, которая является биологически эквивалентной препарату, имеющему более чем 95% абсолютную биологическую доступность и Cmax от 800 до 820 нг/л, где биологическая эквивалентность устанавливается с помощью:
a) 90% доверительного интервала для AUC, который находится в пределах между 80 и 125%, и
b) 90% доверительного интервала для Cmax, который находится в пределах между 80 и 125%.
6. Капсула по п.1, в которой указанная композиция внутренней начинки содержит кислород в количестве, которое опосредует не более чем примерно 3,0 мас.% окислительной деградации, когда указанная дозированная форма хранится в течение трех месяцев при 40°С и 75% RH.
7. Капсула по п.1, в которой не менее чем примерно 75% указанного палоносетрона или его фармацевтически приемлемой соли растворяется через 45 мин, когда исследуется в устройстве для растворения типа II с лопастной мешалкой в соответствии с Фармакопеей США при 75 об./мин и 37°С в 500 мл 0,01н. HCl.
8. Капсула по п.1, в которой не менее чем примерно 75% указанного палоносетрона или его фармацевтически приемлемой соли растворяется через 30 мин, когда исследуется в устройстве для растворения типа II с лопастной мешалкой в соответствии с Фармакопеей США при 75 об./мин и 37°С в 500 мл 0,01н. HCl.
9. Капсула по п.1, в которой указанная оболочка имеет проницаемость для кислорода, меньшую чем примерно 1,0´10-4 мл×см/(см2×24 ч×атм).
10. Капсула по п.1, в которой указанная внутренняя начинка содержит от 0,5 до 4 мас.% поверхностно-активного вещества.
11. Дозированная форма по п.1, в которой указанный палоносетрон или его фармацевтичеки приемлемая соль содержит (3S)-3-[(3aS)-1-оксо-2,3,3а,4,5,6-гексагидро-1H-бензо[de]изохинолин-2-ил]-1-азониабицикло[2.2.2]октан-1-олат или ее фармацевтически приемлемую соль в количестве меньшем, чем 1,0 мас.% по отношению к массе указанного палоносетрона, и где указанная композиция внутренней начинки содержит кислород в количестве, которое опосредует не более чем примерно 3,0 мас.% окислительной деградации указанного палоносетрона или его фармацевтически приемлемой соли, когда указанная дозированная форма хранится три месяца или больше при 40°С и 75% RH.
12. Капсула по п.11, демонстрирующая фармакокинетику при пероральном приеме в голодном состоянии, которая является биологически эквивалентной препарату, имеющему более чем 90% абсолютную биологическую доступность, где биологическая эквивалентность устанавливается с помощью 90% доверительного интервала для AUC, который находится в пределах между 80 и 125%.
13. Капсула по п.11, в которой указанная композиция внутренней начинки содержит от примерно 1 мас.% до примерно 20 мас.% воды.
14. Капсула по п.11, демонстрирующая фармакокинетику при пероральном приеме в голодном состоянии, которая является биологически эквивалентной препарату, имеющему более чем 95% абсолютную биологическую доступность, и Cmax от 800 до 820 нг/л, где биологическая эквивалентность устанавливается с помощью:
a) 90% доверительного интервала для AUC, который находится в пределах между 80 и 125%, и
b) 90% доверительного интервала для Cmax, который находится в пределах между 80 и 125%.
15. Капсула по п.11, в которой не менее чем примерно 75% указанного палоносетрона или его фармацевтически приемлемой соли растворяется через 45 мин, когда исследуется в устройстве для растворения типа II с лопастной мешалкой в соответствии с Фармакопеей США при 75 об./мин и 37°С в 500 мл 0,01н. HCl.
16. Капсула по п.11, в которой указанная оболочка имеет проницаемость для кислорода, меньшую чем примерно 1,0´10-4 мл×см/(см2×24 ч×атм).
17. Капсула по п.11, в которой:
a) указанная композиция внутренней начинки содержит глицерин и
b) указанная оболочка содержит глицерин.
18. Капсула по п.11, в которой указанная оболочка содержит желатин, целлюлозу, крахмал или НРМС.
19. Способ оптимизации биологической доступности и стабильности палоносетрона в желатиновой капсуле, содержащей палоносетрон по п.1, включающий в себя:
a) создание мягкой желатиновой наружной оболочки, имеющей проницаемость для кислорода менее чем примерно 1,0´10-3 мл×см/(см2×24 ч×атм); и
b) получение композиции начинки с помощью стадий, включающих в себя:
i) получение от примерно 0,05 до примерно 2,0 мг палоносетрона в виде палоносетрона гидрохлорида, где указанный палоносетрон содержит (3S)-3-[(3aS)-1-оксо-2,3,3а,4,5,6-гексагидро-1H-бензо [de]изохинолин-2-ил]-1-азониабицикло [2.2.2] октан-1-олат в количестве, меньшем чем 3,0 мас.%;
ii) растворение или диспергирование указанного палоносетрона в воде с образованием водного премикса;
iii) смешивание указанного водного премикса с одним или несколькими липофильными наполнителями, при массовом отношении водного премикса к липофильным наполнителям меньше чем 30:70, с образованием перемешанной или гомогенной композиции липофильной начинки;
iv) смешивание поверхностно-активного вещества с указанной водой, указанным водным премиксом или указанной композицией начинки; и
v) балансирование количеств поверхностно-активного вещества и воды в указанной композиции начинки для облегчения биологической доступности палоносетрона из указанной желатиновой капсулы при пероральном приеме и для сведения к минимуму степени деградации палоносетрона; и
c) заполнение указанной наружной оболочки указанной композицией начинки.
20. Способ по п.19, в котором указанная композиция начинки содержит от примерно 0,1 до примерно 10,0 мас.% поверхностно-активного вещества и от примерно 0,1 до примерно 20 мас.% воды.
21. Способ по п.19, в котором указанная композиция начинки содержит от примерно 0,5 до примерно 4 мас.% поверхностно-активного вещества и от примерно 1 до примерно 10 мас.% воды.
22. Способ по п.19, в котором указанная наружная оболочка капсулы дополнительно содержит глицерин, дополнительно включающий в себя смешивание указанного водного премикса с глицерином до или после образования указанной композиции липофильной начинки.
23. Способ получения набора дозированных форм палоносетрона, имеющих пониженное количество примесей и продуктов деградации, опосредуемых кислородом, включающий в себя:
a) смешивание палоносетрона гидрохлорида и одного или нескольких фармацевтически приемлемых наполнителей с образованием смеси по п.1;
b) переработку указанной смеси в множество конечных дозированных форм по п.1; и
c) исследование одной или нескольких указанных конечных дозированных форм на одно или несколько соединений, родственных палоносетрону, выбранных из (3S)-3-[(3aS)-1-оксо-2,3,3а,4,5,6-гексагидро-1H-бензо[de]изохинолин-2-ил]-1-азониабицикло[2.2.2]октан-1-олата, 2-[(3S)-1-азобицикло[2.2.2]окт-3-ил]-2,4,5,6-тетрагидро-2H-бензо[de]изохинолин-1-он гидрохлорида и (3aR)-2-[(S)-1-азобицикло[2.2.2]окт-3-ил]-2,3,3а,4,5,6-гексагидро-1-оксо-1H-бенз[de]изохинолин гидрохлорида или их гидрохлоридной соли.
24. Способ по п.23, включающий в себя исследование на (3S)-3-[(3aS)-1-оксо-2,3,3а,4,5,6-гексагидро-1H-бензо[de]изохинолин-2-ил]-1-азониабицикло[2.2.2]октан-1-олат или его гидрохлоридную соль.
25. Способ по п.23, включающий в себя исследование на 2-[(3S)-1-азобицикло[2.2.2]окт-3-ил]-2,4,5,6-тетрагидро-1H-бензо[de]изохинолин-1-он гидрохлорида или его гидрохлоридную соль.
26. Способ по п.23, включающий в себя исследование на (3aR)-2-[(S)-1-азобицикло[2.2.2]окт-3-ил]-2,3,3а,4,5,6-гексагидро-1-оксо-1H-бенз[de]изохинолин гидрохлорида или его гидрохлоридную соль.
27. Способ по п.23, дополнительно включающий в себя исследование указанного палоносетрона гидрохлорида или указанной конечной дозированной формы на одно или несколько соединений, выбранных из (3S)-3-(1-оксо-2,4,5,6-тетрагидро-2H-бензо[de]изохинолин-2-ил)-1-азониабицикло[2.2.2]октан-1-олата, (3aR)-2-[(R)-1-азобицикло-[2.2.2]окт-3ил]-2,3,3а,4,5,6-гексагидро-1-оксо-2H-бенз[de]изохинолин гидрохлорида, (3aS)-2-[(R)-1-азобицикло[2.2.2]окт-3ил]-2,3,3а,4,5,6-гексагидро-1-оксо-2H-бенз[de]изохинолин гидрохлорида или (3aS)-2-[(S)-1-азобицикло[2.2.2]окт-3ил]-2,3,3а,4,5,6-гексагидро-1-оксо-2H-бенз[de] изохинолин гидрохлорида или их гидрохлоридной соли.

Текст
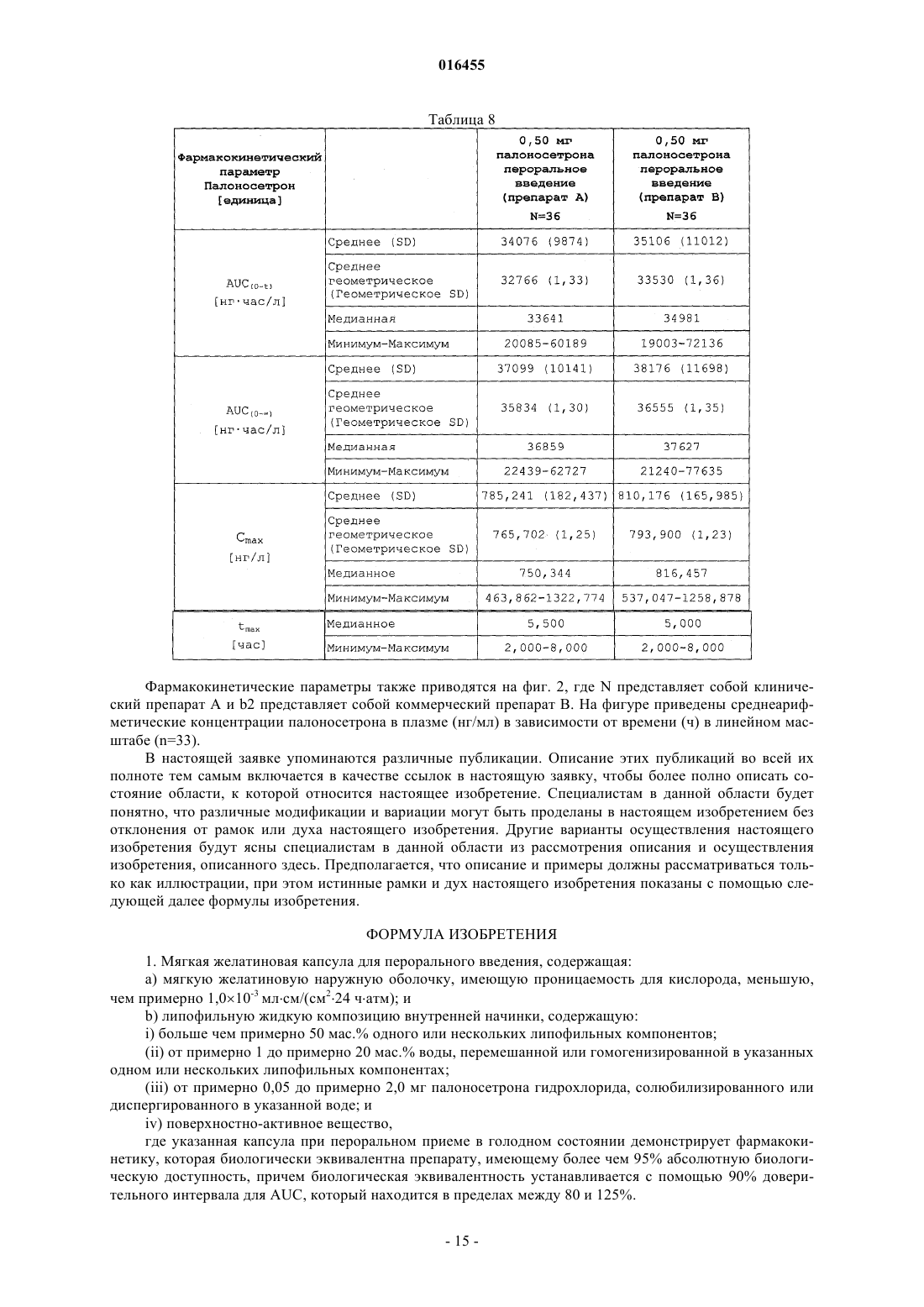
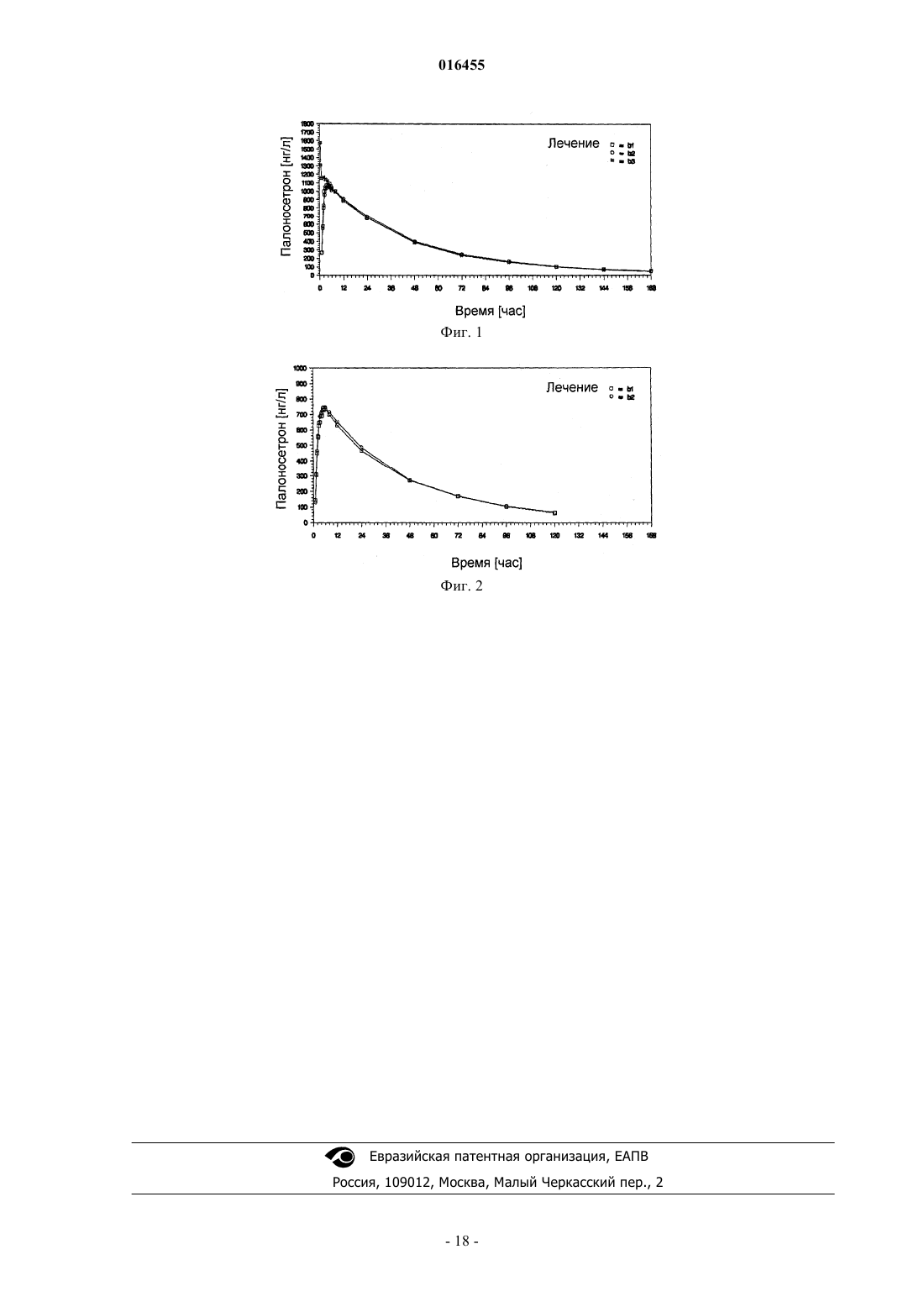
Предусматриваются твердые пероральные дозированные формы палоносетрона гидрохлорида,способы применения дозированных форм для лечения рвоты и способы получения дозированных форм. Дозированные формы имеют улучшенную стабильность и биологическую доступность и предпочтительно находятся в форме капсул, заполненных жидкостью. 016455 Связь с предыдущими заявками Настоящая заявка испрашивает приоритет предварительной заявки на патент Соединенных Штатов 60/854342, зарегистрированной 24 октября 2006 года (срок действия закончился). Область техники, к которой относится изобретение Настоящее изобретение относится к палоносетрону и, в частности, к твердым пероральным дозированным формам палоносетрона гидрохлорида, которые удовлетворяют строгим требованиям стабильности при хранении в упакованном виде. Уровень техники Побочные воздействия тошноты и рвоты при противораковой химиотерапии и радиационной терапии представляют собой широко распространенную и долговременную проблему. Возможно менее известными, но не менее важными, являются послеоперационная тошнота и рвота, которые могут иметь физиологические механизмы, родственные с воздействиями, наблюдаемыми при химиотерапии. Палоносетрона гидрохлорид получен недавно в качестве очень эффективного средства против тошноты и против рвоты для использования вместе с эметогенной противораковой химиотерапией. (Macciocchi, A., ethighly emetogenic chemotherapy-induced nausea and vomiting," Proc. Am. Soc. Clin. Oncol., 2002; Abstract 1480). Палоносетрон также предотвращает послеоперационную тошноту и рвоту. (Chelly, J., et al., "OralRS-25259 prevents postoperative nausea and vomiting following laparoscopic surgery," Anesthesiol., 85(Suppl. 21): abstract no.3 (1996. Способы лечения вызываемой химиотерапией тошноты и рвоты (CINV) и вызываемой радиацией тошноты и рвоты (RINV) с помощью палоносетрона описываются в публикации РСТ заявки на Международный патент WO 2004/045615 от Helsinn Healthcare SA. Способы лечения послеоперационной тошноты и рвоты (PONV) с помощью палоносетрона описаны в публикации РСТ 2004/073714, также от Helsinn Healthcare SA. Палоносетрон является селективным, показывает высокое сродство в качестве антагониста к предшественнику рецептора 5-гидроксилтриптамина 3 (рецептор 5-НТ 3) и показывает низкое сродство к другим рецепторам, таким как допаминовые рецепторы (Wong, E.H.F., et al., "The interaction of RS 25259-197,a potent and selective antagonist, with 5-HT3 receptors, in vitro," Br. J. Pharmacol, 114:851-859 (1995); Eglen,R.M., et al., "Pharmacological characterization of RS 25259-197, a potent and selective antagonist, with 5-HT3receptors, in vivo," Br. J. Pharmacol, 114:860-866 (1995. Палоносетрон представляет собой синтетическое соединение, существующее как единственный изомер, и вводится как гидрохлоридная соль, как представлено в следующей структурной формуле: Официальное химическое наименование для лекарственного средства представляет собой (3aS)-2[(S)-1-азабицикло[2,2,2]окт-3-ил]-2,3,3 а,4,5,6-гексагидро-1-оксо-2H-бенз[de]изохинолина гидрохлорид(CAS119904-90-4); его эмпирическая формула представляет собой C19H24N2OHCl, и его молекулярная масса равна 332,87. Способы синтеза соединения описаны в патентах США 5202333 и 5510486. Палоносетрона гидрохлорид продается в Соединенных Штатах как стерильная жидкость для инъекций, как ALOXI, MGI Pharma и Helsinn Healthcare SA. Жидкость для внутривенных инъекций является прозрачной, бесцветной, непирогенной, находится в изотоническом, буферном растворе. Стабильный изотонический раствор палоносетрона для инъекций описывается в публикации РСТ заявки на Международный патент WO 2004/067005, Helsinn. Несмотря на многочисленные клинические выгоды и преимущества этого препарата для внутривенных инъекций, как правило, наблюдается, что системы доставки лекарственных средств с помощью инъекций доставляют конкретные проблемы относительно времени жизни при хранении и стабильности активного агента при хранении. Они также являются неудобными, когда вводятся самостоятельно, и имеют повышенный риск загрязнения и человеческой ошибки. Таким образом, возможность пероральной доставки для палоносетрона, особенно в твердой форме, была бы особенно привлекательной. Способы улучшения стабильности и времени хранения в упаковке для препаратов палоносетрона также были бы желательными. Сущность изобретения Разрабатываются мягкие гелевые капсулы для палоносетрона, которые демонстрируют превосходную биологическую доступность при пероральном приеме и стабильность, когда хранятся в течение продолжительных периодов времени. Наружная оболочка капсулы основывается на желатине, и внутренняя начинка капсулы представляет собой сплошную липофильную внутреннюю фазу, которая содержит палоносетрон, растворенный в водном компоненте, перемешиваемом или гомогенизируемом в липофильной фазе с помощью минимальных количеств поверхностно-активного вещества. Препарат представляет элегантное решение для устранения несовместимости, наблюдаемой повсеместно между:-1 016455 водной начинкой и стабильностью желатина; деградацией поверхностно-активного вещества и палоносетрона и стабильностью палоносетрона и концентрациейпалоносетрона. По этой причине в первом главном варианте осуществления настоящее изобретение предусматривает мягкую желатиновую капсулу для перорального введения, содержащую: (а) наружную оболочку из мягкого желатина, имеющую проницаемость для кислорода, меньшую, чем примерно 1,010-3 млсм/(см 224 чатм); и (b) композицию внутренней начинки из липофильной жидкости, содержащую: (i) больше, чем примерно 50 мас.% одного или нескольких липофильных компонентов; (ii) от примерно 1 до примерно 20 мас.% воды, перемешанной или гомогенизированной в указанном одном или нескольких липофильных компонентах; (iii) от примерно 0,05 до примерно 2,0 мг палоносетрона в виде палоносетрона гидрохлорида, солюбилизированного или диспергированного в указанной воде; и (iv) от примерно 0,5 до примерно 5 мас.% поверхностно-активного вещества. Разрабатываются также препараты и способы получения, которые могут определяться по количеству или по концентрации палоносетрона в дозированной форме и по побочным продуктам деградации в дозированной форме. Один из таких побочных продуктов деградации представляет собой продукт деградации, опосредуемый кислородом, и упоминается здесь как "Cpd1". Также разрабатываются дозированные формы палоносетрона, включая способы получения, с повышенной стабильностью, благодаря их защите от кислорода и опосредуемой кислородом деградации. На основе этих открытий и разработок, разрабатываются дозированные формы, которые могут определяться по одному или нескольким из следующих физических признаков: оболочка или покрытие, которые, по существу, непроницаемы для кислорода; использование жидкой начинки внутри оболочки капсулы, предпочтительно содержащей воду; минимальное содержание кислорода в жидкой начинке; химические средства для предотвращения окислительной деградации; стойкая к влажности упаковка, которая является стойкой к проникновению кислорода; и/или использование окружающей среды, обедненной кислородом, при получении дозированных форм. Эти дозированные формы имеют превосходную стабильность в течение продолжительных периодов времени, превосходную стойкость к окислительной деградации и превосходную биологическую доступность при пероральном приеме. Эти дозированные формы могут использоваться при лечении любого заболевания, для которого палоносетрон имеет клиническое применение, но предпочтительно они используются для лечения рвоты. Во втором главном варианте осуществления, по этой причине, настоящее изобретение предусматривает дозированную форму капсулы для перорального введения, содержащую: (а) наружную оболочку,имеющую проницаемость для кислорода меньше, чем примерно 1,010-3 млсм/(см 224 чатм); и (b) композицию внутренней начинки, содержащую: от примерно 0,05 до примерно 2,0 мг палоносетрона, в виде палоносетрона гидрохлорида, где указанный палоносетрон содержит Cpd1 в количестве, меньшем, чем 1,0 мас.%; при этом не более чем 5,0 мас.% указанного палоносетрона гидрохлорида деградирует, когда указанная дозированная форма хранится три месяца или больше при 40 С и 75% RH (относительной влажности). Разумеется, настоящее изобретение могло бы осуществляться с использованием дозированных форм, иных, чем капсулы, и другой вариант осуществления настоящего изобретения предусматривает твердую пероральную дозированную форму, содержащую: (а) наружную оболочку или покрытие,имеющее проницаемость для кислорода меньше, чем примерно 1,010-3 млсм/(см 224 чатм); и (b) композицию внутренней начинки, содержащую: от примерно 0,05 до примерно 2,0 мг палоносетрона, в виде палоносетрона гидрохлорида, где указанный палоносетрон содержит Cpdl в количестве, меньшем, чем 1,0 мас.%; где не более чем 5,0 мас.% указанного палоносетрона гидрохлорида деградирует, когда указанная дозированная форма хранится в течение трех месяцев или больше при 40 С и 75% RH. Также разрабатываются способы получения дозированных форм палоносетрона, которые имеют пониженное количество примесей и продуктов деградации, опосредуемых кислородом, и дозированных форм палоносетрона, получаемых с помощью этих способов. Таким образом, еще в одном варианте осуществления, настоящее изобретение предусматривает способ получения набора дозированных форм палоносетрона, имеющих пониженное количество примесей и продуктов деградации, опосредуемых кислородом, включающий в себя (а) смешивание палоносетрона гидрохлорида и одного или нескольких фармацевтически приемлемых наполнителей, с образованием смеси; (b) переработку указанной смеси в виде множества конечных дозированных форм и (с) исследование одной или нескольких из указанных конечных дозированных форм на Cpd1. Этот способ может осуществляться с помощью любой дозированной формы, включая капсулу, гелькап или ампулу, заполненную жидкостью. Дополнительные преимущества настоящего изобретения будут частично представлены в описании,которое следует далее, а частично станут очевидны из описания, или они могут быть изучены при осуществлении настоящего изобретения. Преимущества настоящего изобретения будут реализованы и получены с помощью элементов и сочетаний, конкретно описанных в прилагаемой формуле изобретения.-2 016455 Необходимо понять, что как предшествующее общее описание, так и следующее далее подробное описание, являются только иллюстративными и пояснительными, а не ограничивающими настоящее изобретение, как описано в формуле изобретения. На фигурах: фиг. 1 представляет собой график фармакокинетики, наблюдаемой у пациентов людей при исследованиях биологической эквивалентности, где b1 представляет собой лечение с помощью клинического препарата А, b2 представляет собой лечение с помощью коммерческого препарата В, и b3 представляет собой лечение с помощью Aloxi, внутривенно; фиг. 2 представляет собой график фармакокинетики, наблюдаемой у пациентов людей при исследованиях биологической эквивалентности, где b1 представляет собой клинический препарат А, а b2 представляет собой коммерческий препарат В. Обе фигуры показывают среднеарифметические концентрации палоносетрона в плазме (нг/мл) в зависимости от времени (ч) в линейном масштабе (n=33). Подробное описание изобретения Настоящее изобретение может быть понято легче посредством ссылок на следующее далее подробное описание предпочтительных вариантов осуществления настоящего изобретения и примеров, содержащихся здесь. Определение и использование терминов Как используется в настоящем описании и в формуле изобретения, которые следуют далее, формы единственного числа включают в себя ссылки на множественное число, если только контекст не диктует четко иного. Так, например, ссылка на "ингредиент" включает в себя смеси ингредиентов, ссылка на "активный фармацевтический агент" включает в себя более одного активного фармацевтического агента, и тому подобное."Лечение" или "излечение" заболевания включает в себя (1) предотвращение возникновения заболевания у животного, которое может быть предрасположено к заболеванию, но еще не испытывает или не проявляет симптомов заболевания, (2) замедление заболевания, то есть приостановку его развития,или (3) ослабление заболевания, то есть осуществление регрессии заболевания. Как здесь используется, непосредственная окружающая среда относится к окружающей среде, непосредственно окружающей элемент или способ, как правило, к газовой среде, с которой элемент или способ находится в контакте и сообщении."Рвота" для целей настоящей заявки будет иметь значение шире, чем обычное словарное определение, и включает в себя не только рвоту, но также тошноту и позывы к рвоте."Умеренно эметогенная химиотерапия" относится к химиотерапии, при которой эметогенный потенциал является сравнимым или эквивалентным с эметогенным потенциалом карбоплатина, цисплатина 50 мг/м 2, циклофосфамида 1500 мг/м 2, доксорубицина 25 мг/м 2, эпирубицина, иринотекана или метотрексата 250 мг/м 2,"В высшей степени эметогенная химиотерапия" относится к химиотерапии, при которой эметогенный потенциал является сравнимым или эквивалентным с эметогенным потенциалом цисплатина 60 мг/м 2, циклофосфамида 1500 мг/м 2 или дакарбазина."Фармацевтически приемлемый" означает, что он является пригодным для использования при получении фармацевтической композиции, которая является в целом безопасной, нетоксичной и не является нежелательной, ни биологически, ни в ином смысле, и включает в себя то, что она является приемлемой для ветеринарного применения, а также для фармацевтического применения на людях."Терапевтически эффективное количество" означает то количество, которое, когда вводится животному для лечения заболевания, является достаточным для осуществления такого лечения заболевания."de minimis" количество кислорода относится к такому количеству кислорода, которое дает возможность для деградации не более чем примерно 0,5, 1,0, 1,5, 2,0, 2,5 или 3,0 мас.% указанного палоносетрона (предпочтительно определяют по деградации до Cpd1), когда хранят при комнатной температуре при условиях окружающей среды в течение шести, двенадцати, восемнадцати, двадцати четырех, тридцати или тридцати шести месяцев. Стабильность при хранении в упаковке для целей настоящего изобретения измеряется посредством хранения дозированной формы в ее упаковке при 40 С, при относительной влажности 75% или при условиях окружающей среды в течение трех, шести, двенадцати, восемнадцати, двадцати четырех, тридцати или тридцати шести месяцев. Стабильный препарат представляет собой такой, в котором не более чем примерно 0,5, 1,0, 1,5, 2,0, 2,5, 3,0, или 5,0 мас.% палоносетрона в дозированной форме деградирует(предпочтительно определяют по деградации до одного или нескольких продуктов деградации, описанных здесь). Когда диапазоны приводятся посредством указания нижнего предела диапазона отдельно от верхнего края диапазона, необходимо понять, что диапазон может определяться посредством селективного объединения любого из переменных нижнего края с любым из переменных верхнего края, которое является математически возможным.-3 016455 Когда здесь используется термин "примерно" или "са.", он будет компенсировать разброс, допустимый для фармацевтической промышленности и присущий фармацевтическим продуктам, такой как различия в прочности продукта из-за разброса при получении и обусловленной временем деградации продукта. Термин делает возможным любой разброс, который в практике фармацевтических препаратов дал бы возможность для оценки продукта как считающегося биологически эквивалентным относительно упоминаемой силе воздействия заявляемого продукта. Термин "абсолютная биологическая доступность" относится к доступности активного лекарственного средства при системной циркуляции после введения, отличного от внутривенного (то есть после перорального, ректального, трансдермального, подкожного введения). Для определения абсолютной биологической доступности лекарственного средства должны осуществляться фармакокинетические исследования для получения графика зависимости концентрации лекарственного средства в плазме от времени для лекарственного средства, как после внутривенного (IV), так и после введения, отличного от внутривенного. Абсолютная биологическая доступность представляет собой скорректированную на дозу площадь под кривой (AUC) для введения, отличного от внутривенного, деленную на AUC для внутривенного введения. О препарате говорится, что он является биологически эквивалентным в терминах абсолютной биологической доступности эталонному препарату, когда имеется установленный 90% доверительный интервал для AUC(0-), которая находится в пределах между 80 и 125%, по отношению к уровню биологической доступности для эталонного препарата. Когда здесь приводятся фармакокинетические параметры (то есть Tmax, абсолютная биологическая доступность, и тому подобное), будет понятно, что они могут относиться к средней медианной или индивидуальной наблюдаемой фармакокинетике, и что в формуле изобретения предполагается средняя фармакокинетика, если не утверждается противоположного. Фармакокинетический параметр также будет пониматься как наблюдаемый в голодном состоянии, если не утверждается противоположного. Обсуждение Как рассмотрено выше, настоящее изобретение предусматривает твердые пероральные дозированные формы, которые имеют улучшенную стабильность и стойкость к окислительной деградации, основывающиеся на нескольких технологиях приготовления, включая использование покрытия или оболочки, которая является, по существу, непроницаемой для кислорода, или использование липофильной жидкой начинки, имеющей воду, гомогенизированную или перемешанную с ней. В первом главном варианте осуществления настоящее изобретение предусматривает твердую пероральную дозированную форму,содержащую: (а) наружную оболочку или покрытие, имеющее проницаемость для кислорода меньшую,чем примерно 1,010-3 млсм/(см 224 чатм); и (b) композицию внутренней начинки, содержащую: от примерно 0,05 до примерно 2,0 мг палоносетрона в виде палоносетрона гидрохлорида, где указанный палоносетрон содержит Cpd1 в количестве менее чем 1,0 мас.%; где указанная дозированная форма демонстрирует стабильность при хранении в упаковке, предпочтительно определяемую так, что не более чем 5,0 мас.% указанного палоносетрона гидрохлорида деградирует, когда указанная дозированная форма хранится три месяца или больше при 40 С и 75% RH. Настоящее изобретение дополнительно предусматривает способ лечения рвоты, включающий в себя пероральное введение пациенту, страдающему от рвоты или имеющего риск пострадать от рвоты, дозированный формы по настоящему изобретению. Настоящее изобретение может осуществляться с помощью любого типа твердой пероральной дозированной формы, определяемой как любая дозированная форма, которая вводится пероральным путем и заглатывается, включая, например, капсулу или гелькап (то есть капсулу, заполненную жидкостью). В предпочтительном варианте осуществления дозированная форма представляет собой капсулу, а еще в более предпочтительном варианте осуществления дозированная форма представляет собой гелькап, заполненный жидкостью. Что не представляла бы собой дозированная форма, она предпочтительно имеет наружную оболочку или покрытие, которое имеет минимальную проницаемость для кислорода. В предпочтительных вариантах осуществления настоящего изобретения, покрытие или оболочка имеет проницаемость для кислорода, которая меньше чем примерно 1,010-3, 5,010-4, 1,010-4, 5,010-5 или даже 2,010-5 млсм/(см 224 чатм). Предпочтительная дозированная форма для настоящего изобретения представляет собой капсулу,имеющую наружную оболочку, которая растворяется в желудочных жидкостях. Заполненная жидкостью капсула, предпочтительно содержащая воду, является особенно предпочтительной благодаря однородности содержимого и дозы при работе с жидкостями и возможности сведения к минимуму экспонирования для кислорода при получении дозированной формы и хранении дозированной формы в течение продолжительных периодов времени. Среди доступных наружных оболочек мягкая наружная оболочка представляет собой предпочтительную структуру оболочки, благодаря ее способности удерживать жидкости и противостоять прохождению кислорода. Предпочтительные материалы для наружной оболочки "гелькап" включают в себя,например, желатин, целлюлозу, крахмал или НРМС (гидроксипропилметилцеллюлозу). В предпочтительном варианте осуществления оболочка содержит желатин и, необязательно, один или несколько на-4 016455 полнителей для оболочки, выбранных из глицерина, сорбитола и двуокиси титана. Жидкая композиция, которая заполняет капсулу, предпочтительно является (1) преимущественно липофильной и (2) присутствующей как непрерывная жидкая фаза (то есть, такая, в которой жидкие компоненты являются либо перемешанными, либо полностью гомогенизированными/эмульсифицированными). Сплошная фаза является предпочтительной благодаря простоте обработки и однородности композиции. Жидкая начинка содержит основу наполнителя и активный агент, равномерно распределенный в жидкой начинке. Кроме того, активный агент предпочтительно является растворенным или диспергированным как микроэмульсия в основе наполнителя. Общая масса композиции начинки предпочтительно является большей, чем примерно 50, 75 или 100 мг, и предпочтительно является меньшей, чем примерно 500, 250, 200 или 150 мг, наиболее предпочтительно находится в пределах от примерно 100 до примерно 150 мг. Жидкая начинка предпочтительно состоит в основном из одного или нескольких липофильных компонентов в количестве от примерно 50 мас.% до примерно 99 мас.% предпочтительно от примерно 75 мас.% до примерно 98 мас.%. Предпочтительные липофильные компоненты включают в себя, например,моно- и диглицериды жирных кислот, в частности, включая в себя моно- и диглицериды каприловой/каприновой кислоты. Жидкая начинка может также содержать глицерин, предпочтительно, в количестве от примерно 1 до примерно 15 мас.%, более предпочтительно от примерно 2 до примерно 10 мас.%. В одном из предпочтительных вариантов осуществления композиция, как оболочки, так и внутренней начинки содержит глицерин. В другом предпочтительном варианте осуществления жидкая начинка содержит 0,25, 0,35 мг или более палоносетрона в виде палоносетрона гидрохлорида (то есть 0,50 или 0,75 мг); солюбилизированного при солюбилизирующем эффективном количестве жидкости, содержащей липофильный наполнитель и воду. Композиция начинки может содержать различные средства для облегчения перехода палоносетрона из дозированной формы в желудочно-кишечные жидкости желудочно-кишечного тракта, так чтобы палоносетрон мог легче поглощаться в кровотоке. Например, композиция жидкой начинки может содержать поверхностно-активное вещество, оптимально, в количестве от примерно 0,1 мас.% до примерно 6 мас.%, от примерно 0,5 мас.% до примерно 5 мас.% или от примерно 1,0 мас.% до примерно 3,0 мас.% Композиция жидкой начинки предпочтительно содержит более чем 0,1, 0,5 или 1,0 мас.% поверхностноактивного вещества и меньше чем 10, 8, 5,4, или даже 4 мас.% поверхностно-активного вещества. Особенно предпочтительное поверхностно-активное вещество представляет собой полиглицерилолеат. Альтернативно или в дополнение к этому, средства переноса для заполненной жидкостью капсулы могут включать в себя воду, которая образует одну фазу или микроэмульсию со всеми другими жидкими ингредиентами в основе наполнителя. Композиция жидкой начинки предпочтительно содержит от примерно 0,05 мас.% до примерно 30 мас.% воды, от примерно 1 мас.% до примерно 20 мас.% воды или от примерно 2 мас.% до примерно 10 мас.% воды. Жидкая начинка предпочтительно содержит более чем 0,1, 0,5 или 1,0 мас.% воды и менее чем 20, 15, 10, 8 или 5 мас.% воды. Кроме того, основа наполнителя может содержать один или несколько химических агентов для предотвращения опосредуемой кислородом деградации палоносетрона в дозированной форме. Например, основа наполнителя может содержать хелатирующий агент, такой как этилендиаминтетрауксусная кислота (EDTA), антиоксидант, такой как бутилированный гидроксианизол (ВНА), или восстанавливающий агент, в количестве, находящемся в пределах от примерно 0,005 мас.% до примерно 2,0 мас.%,более предпочтительно от примерно 0,01 мас.% до примерно 1,0 мас.% или от примерно 0,05 мас.% до примерно 0,5 мас.%. В предпочтительном варианте осуществления основа наполнителя содержит антиоксидант. Активный агент, который предпочтительно представляет собой палоносетрон гидрохлорид, предпочтительно присутствует в композиции начинки в количестве, находящемся в пределах от примерно 0,01 до примерно 10,0 мас.%, от примерно 0,05 до примерно 5,0 мас.% или от примерно 0,1 до примерно 2,0 мас.%. Альтернативно, обнаружены особенно стабильные препараты, где концентрация палоносетрона превышает 0,3%, предпочтительно он находится при концентрации не больше чем примерно 1 мас.%. Особенно важной особенностью композиции внутренней начинки, которая является предпочтительной в любом из вариантов осуществления настоящего изобретения вне зависимости от дозированной формы или типа начинки или способа получения, является минимальное содержание кислорода. В предпочтительном варианте осуществления, композиция внутренней начинки содержит кислород в таком количестве, при котором деградирует не более чем примерно 3,0, 2,5, 2,0, 1,5, 1,0 или 0,5 мас.% указанного палоносетрона, когда дозированная форма хранится в режимах исследования стабильности при хранении в упаковке, например, в течение трех месяцев при 40 С и 75% RH. Это количество предпочтительно измеряют с помощью количества Cpd1 в композициях. Другой важный признак препаратов по настоящему изобретению представляет собой их фармакокинетику. Определено, что дозированные формы по настоящему изобретению имеют абсолютную биологическую доступность приблизительно 100%, в пределах биологической эквивалентности. Таким образом, например, в то время как 0,75 мг инъекция палоносетрона дает среднюю AUC(0-) са. 58285(нгч/л), 0,75 мг гелькап дает среднюю AUC(0-) ca. 57403 (нгч/л). В противоположность этому, средняяCmax для 0,75 мг гелькапа составляет примерно 1224 нг/л, в то время как 0,75 мг инъекция дает среднююCmax примерно 1665 нг/л. 0,50 мг гелькапа, как показано, дает среднюю AUC(0-) ca. 38176 (нгч/л), и среднюю Cmax примерно 810 нг/л, тем самым демонстрируя фармакокинетику, пропорциональную дозе. В различных вариантах осуществления, следовательно, дозированная форма по настоящему изобретению дает больше чем 90, 95 или даже 98% абсолютную биологическую доступность, как среднеарифметическое значение, опять же, в пределах биологической эквивалентности. Альтернативно или в дополнение к этому, 50 мг гелькап дает среднюю Cmax от примерно 700 до примерно 950 нг/мл, или от примерно 750 до примерно 875 нг/мл. В наиболее предпочтительном варианте осуществления, 50 мг гелькап дает Cmax от 800 до 820 нг/л, предпочтительно в пределах биологической эквивалентности. Поскольку дозированные формы по настоящему изобретению демонстрируют фармакокинетику, пропорциональную дозе, будет понятно, что эти значения Cmax могут стандартизоваться на основе силы воздействия дозированной формы и что значения Cmax могут приписываться альтернативным значениям силы воздействия на основе такой стандартизации. Еще один важный признак дозированных форм по настоящему изобретению, который также является предпочтительным в любом из вариантов осуществления настоящего изобретения, относится к растворению дозированной формы, а в предпочтительном варианте осуществления, не менее чем примерно 75% палоносетрона в дозированной форме растворяется через 30 или 45 мин, когда исследуется в устройстве для растворения типа II с лопастной мешалкой в соответствии с Фармакопеей США, при 75 об/мин и 37 С, в 500 мл, 0,01 н. HCl. Еще один признак дозированных форм по настоящему изобретению, который также является предпочтительным в любом из вариантов осуществления настоящего изобретения, вне зависимости от дозированной формы или типа начинки или способа получения, заключается в том, что дозированная форма испытывает деградацию палоносетрона не более чем 5, 3 или 2 мас.%, когда дозированная форма в ее стойкой к влажности упаковке экспонируется для окружающей среды 25 С и 60% RH, или 40 С и 75%RH, в течение периодов, равных или превышающих 3 месяц, шесть месяцев, 9 месяцев или даже один год. Палоносетрона гидрохлорид и родственные соединения Палоносетрон, используемый в настоящем изобретении, может представлять собой палоносетрон в качестве основания или фармацевтически приемлемой соли, но предпочтительно представляет собой палоносетрона гидрохлорид. В дополнение к этому, палоносетрон предпочтительно присутствует в количестве, находящемся в пределах от примерно 0,02 мг до примерно 10 мг на дозированную форму, более предпочтительно от примерно 0,05 или 0,15 до примерно 2 мг на дозированную форму, и еще более предпочтительно от примерно 0,2 до примерно 1,0 мг на дозированную форму, по отношению к массе основы, когда присутствует в качестве фармацевтически приемлемой соли. Особенно предпочтительные дозы представляют собой 0,25 мг, 0,50, и 0,75 мг палоносетрона или его соли по отношению к массе основы. Особенно стабильные препараты обнаружены посредством использования количеств палоносетрона в жидких гелькапах, больших чем примерно 0,25, 0,35 или 0,45 мг, предпочтительно меньших чем примерно 2,0 мг. Палоносетрона гидрохлорид, используемый для получения дозированной формы или содержащийся в конечной дозированной форме, может также отличаться присутствием различных родственных к палоносетрону соединений, включая соединения Cpd3, Cpd2 и/или Cpd1, как описывается с помощью следующих далее химических структур: Соединения Cpd2 и Cpd3, как правило, присутствуют, индивидуально или в сочетании по отношению к палоносетрона гидрохлориду, в количествах, меньших чем 1,0, 0,75, или 0,5 мас.% и/или больших,чем примерно 0,05, 0,075 или 0,1 мас.% Cpd2 и Cpd3 могут измеряться в дозированной форме или в сыром материале палоносетрона, используемом для получения дозированной формы. Соединение Cpd1, как правило, присутствует, индивидуально по отношению к палоносетрона гидрохлориду, в количестве,большем чем примерно 0,05, 0,1 или 0,2 мас.% и/или меньшем чем примерно 3,0, 2,5, 2,0, 1,5, 1,0 или 0,5-6 016455 мас.% Cpd1 предпочтительно измеряется в дозированной форме, поскольку это мера опосредуемой кислородом деградации. В одном из предпочтительных вариантов осуществления, дозированные формы определяются по стабильности, при которой не более чем примерно 5,0, 4,0, 3,0, 2,5, 2,0, 1,5, 1,0 или 0,5 мас.% соединения Cpd1 образуется, когда дозированная форма в ее упаковке, стойкой к влажности, экспонируется для окружающей среды при 25 С и 60% RH или при 40 С и 75% RH в течение периодов,равных или превышающих 3 месяца, 6 месяцев, 9 месяцев или даже один год. По этой причине, в другом варианте осуществления настоящее изобретение предусматривает твердую пероральную дозированную форму, содержащую: (а) от примерно 0,05 до примерно 2,0 мг палоносетрона или его фармацевтически приемлемой соли; (b) один или несколько фармацевтически приемлемых наполнителе; (с) Cpd1 в количестве, меньшем чем 3,0 мас.% по отношению к массе палоносетрона. В другом варианте осуществления настоящее изобретение предусматривает твердую пероральную дозированную форму, содержащую: (а) от примерно 0,05 до примерно 2,0 мг палоносетрона или его фармацевтически приемлемой соли; (b) один или несколько фармацевтически приемлемых наполнителей; (с)Cpd2 или Cpd3, в количестве, меньшем, чем 1,0 мас.% по отношению к массе палоносетрона или его фармацевтически приемлемой соли. В любом из этих варианты осуществления, дозированная форма может необязательно содержать средства для предотвращения опосредуемой кислородом деградации указанного палоносетрона. Другие родственные палоносетрону соединения, которые могут присутствовать в композициях,включают в себя Cpd4, Cpd5, Cpd6 и Cpd7, как изображено ниже: Способы получения Настоящее изобретение также предусматривает способы получения дозированных форм палоносетрона. Таким образом, еще в одном варианте осуществления настоящее изобретение предусматривает способ получения набора дозированных форм палоносетрона, имеющих пониженное количество примесей и продуктов деградации, опосредуемых кислородом, включающий в себя (а) смешивание палоносетрона гидрохлорида и одного или нескольких фармацевтически приемлемых наполнителей с образованием смеси; (b) переработку указанной смеси в виде множества конечных дозированных форм и (с) исследование одной или нескольких из указанных конечных дозированных форм на одно или несколько соединений, родственных палоносетрону, выбранных из Cpd2, Cpd1 и Cpd3. "Переработка" относится к стадиям, используемым для получения фармацевтического препарата и конечной дозированной формы из определенного набора ингредиентов, и исключает способы химического синтеза ингредиентов, используемых в препарате. Этот вариант осуществления распространяется на все дозированные формы палоносетрона, включая ампулы со стандартными единичными дозами, заполненные палоносетроном,например, вместе со стерильной жидкостью для инъекций. Таким образом, например, настоящее изобретение может распространяться на способы заполнения ампул или контейнеров для стандартных единичных доз стерильными растворами палоносетрона для инъекций, предпочтительно в водных средах и предпочтительно приготовленных, как описано в заявке на международный патент WO 2004/067005,Calderari et al. В этом контексте "ампула" обозначает малый герметичный контейнер медицинского препарата, который используют только один раз и который включает в себя разламываемые и неразламываемые стеклянные ампулы, разламываемые пластиковые ампулы, миниатюрные баночки с крышкой на резьбе и любой другой тип контейнера с размером, пригодным для удерживания только одной стандартной единичной дозы палоносетрона (как правило, примерно 5 мл). Другой вариант осуществления охватывает баланс, достигаемый с помощью препаратов по настоящему изобретению, по отношению к биологической доступности и стабильности, и в этом варианте осуществления настоящее изобретение предусматривает способ оптимизации биологической доступности и стабильности палоносетрона в желатиновой капсуле палоносетрона, включающий в себя: (а) создание мягкой желатиновой наружной оболочки, имеющей проницаемость для кислорода меньше, чем примерно 1,010-3 м 2 млсм/(см 224 чатм); и (b) получение композиции начинки с помощью стадий, включающих в себя: (i) создание от примерно 0,05 до примерно 2,0 мг палоносетрона в виде палоносетрона гидрохлорида, где указанный палоносетрон содержит Cpd1 в количестве, меньшем чем 1,0 мас.% по отношению к массе указанного палоносетрона; (ii) растворение или диспергирование указанного палоносе-7 016455 трона в воде с образованием водного премикса; (iii) смешивание указанного водного премикса с одним или несколькими липофильными наполнителями, при массовом отношении водного премикса к липофильным наполнителям, меньшем, чем 50:50, 40:60, 30:70 или 20:80, с образованием перемешанной или гомогенной липофильной композиции начинки; (iv) смешиванием поверхностно-активного вещества с указанной водой, указанным водным премиксом или указанной композицией начинки, и (v) балансирование количеств поверхностно-активного вещества и воды в указанной композиции начинки для облегчения биологической доступности палоносетрона из указанной желатиновой капсулы при пероральном приеме и для сведения к минимуму степени деградации палоносетрона; и (с) заполнение указанной наружной оболочки указанной композицией начинки. Еще один способ по настоящему изобретению включает в себя способ упаковки дозированной формы палоносетрона, включающий в себя: (а) создание пустой оболочки; и (b) заполнение указанного контейнера в форме оболочки композицией начинки в непосредственной окружающей среде, обедненной кислородом, где указанная композиция начинки содержит: (i) определенное количество композиции активного ингредиента, содержащего палоносетрон или его фармацевтически приемлемую соль; и (ii) фармацевтически приемлемый наполнитель. "Окружающая среда, обедненная кислородом" предпочтительно представляет собой среду, определяемую с помощью содержания кислорода меньше, чем примерно 10% кислорода, 5% кислорода или даже 1 или 0,1% кислорода (по массе или по объему). В еще более предпочтительном варианте осуществления способы получения или упаковки дозированных форм по настоящему изобретению осуществляют в атмосфере азота или с продувкой азотом в обогащенной азотом окружающей среде, содержащей более чем примерно 90, 95 или 98% азота (по массе или по объему). В другом конкретном варианте осуществления, способ определяется вариабельностью активного ингредиента среди дозированных форм, при этом предусматривается способ получения множества твердых пероральных дозированных форм, включающий в себя: (а) создание оболочки капсулы; (b) заполнение указанной оболочки композицией начинки, содержащей: (i) определенное количество палоносетрона или его фармацевтически приемлемой соли и (ii) фармацевтически приемлемый наполнитель; и (с) повторение стадий (а) и (b) дополнительно один или несколько раз, где указанное определенное количество имеет разброс от одной капсулы до другой, меньший, чем примерно 3, 2, 1, 0,5 или 0,1 мас.%. В любом из указанных выше вариантов осуществления способ получения может также дополнительно включать в себя упаковку указанной дозированной формы или множества дозированных форм в стойкий к влажности герметичный контейнер. Материал, используемый для получения стойкого к влажности герметичного контейнера, предпочтительно имеет проницаемость для кислорода меньше, чем примерно 1,010-2, 1,010-3, 1,010-4, или даже 5,010-5 млсм/(см 224 чатм). Альтернативно или в дополнение к этому, упаковка может характеризоваться как "плотный контейнер" согласно стандартам,описанным в Фармакопее США 671 (то есть не более одного из десяти исследуемых контейнеров превышают проницаемость для влажности 100 мг в день на л, и ни один не превышает 200 мг в день на мл) . Кроме того, контейнер может определяться по количеству влажности, которое он позволяет поглотить дозированным формам по настоящему изобретению во время хранения. Например, в различных предпочтительных вариантах осуществления контейнер предотвращает поглощение указанными дозами более чем 1,0, 0,1 или даже 0,05 мас.% влажности через три месяца при хранении при 40 С и 75% относительной влажности. Блистерная упаковка является особенно предпочтительным способом упаковки. Мягкие желатиновые капсулы Фармацевтические композиции с жидкой сердцевиной по настоящему изобретению инкапсулируют в мягкую желатиновую оболочку, описанную ниже. Желатин представляет собой предпочтительный компонент мягких желатиновых оболочек по настоящему изобретению. Исходный желатиновый материал может быть получен с помощью частичного гидролиза коллагенового материала, такого как кожа,белые соединительные ткани или кости животных. Желатиновый материал может классифицироваться как желатин типа А, который получают посредством кислотной обработки свиной кожи и который демонстрирует изоэлектрическую точку в пределах между рН 7 и рН 9; и желатин типа В, который получают от щелочной обработки костей и кож животных (коров), и который демонстрирует изоэлектрическую точку в пределах между рН 4,7 и рН 5,2. Смеси желатинов типа А и типа В могут использоваться для получения желатина с необходимыми характеристиками вязкости и прочности по Блуму для получения капсулы. Желатин, пригодный для получения капсулы, является коммерчески доступным от SigmaChemical Company, St. Louis, Mo. Относительно общего описания желатина и капсул на основе желатина,см. Remington's Pharmaceutical Sciences, 16th ed., Mack Publishing Company, Easton, Pa (1980), page 1245 иpages 1576-1582; и патент США 4935243, Borkan et al., выдан 19 июня 1990 года; эти два источника включаются сюда в качестве ссылок во всей их полноте. Мягкие желатиновые оболочки могут содержать от примерно 20% до примерно 60% желатина. Желатин может быть типа А или типа В или представлять собой их смесь, с числами Блума, находящимися в пределах от примерно 60 до примерно 300. Мягкие желатиновые оболочки могут также содержать пластификатор. Пригодные для использования пластификаторы включают в себя глицерин, сорбитан, сорбитол или сходные низкомолекулярные полиолы и их смеси. Предпочтительный пластификатор, пригодный для использования в настоящем изобретении, представляет собой глицерин. Мягкие желатиновые-8 016455 оболочки по настоящему изобретению могут также содержать воду. Не ограничиваясь теорией, вода, как предполагается, помогает в быстром растворении или разрыве мягкой желатиновой оболочки при контакте с желудочно-кишечными жидкостями, встречающимися в организме. Мягкие желатиновые капсулы и способы инкапсулирования описаны в Р. К. Wilkinson et al., "Softgels: Manufacturing Considerations", Drugs and Pharmaceutical Sciences, 41 (Specialized Drug Delivery Systems), P. Tyle, Ed. (Marcel Dekker, Inc., New York, 1990) pp.409-449; F.S. Horn et at., "Capsules, Soft", Encyclopedia of Pharmaceutical Technology, vol. 2, J. Swarbrick and J.C. Boylan, eds. (Marcel Dekker, Inc., NewEbert, "Soft Elastic Gelatin Capsules: A Unique Dosage Form", Pharmaceutical Technology, vol. 1, no. 5, pp. 44-50 (1977); эти источники включаются сюда в качестве ссылок во всей их полноте. Полученная мягкая желатиновая капсула является растворимой в воде и в желудочно-кишечной жидкости. При заглатывании капсулы желатиновая оболочка быстро растворяется или разрывается в желудочно-кишечном тракте, тем самым вводя активные фармацевтические препараты из жидкой сердцевины в организм. Способы лечения В других вариантах осуществления настоящее изобретение предусматривает способы лечения рвоты посредством введения одной или нескольких дозированных форм, описанных здесь. Рвота может представлять собой рвоту в острой фазе (то есть рвоту, испытываемую в пределах примерно 24 ч после события, вызывающего рвоту) или замедленную рвоту (то есть рвоту, испытываемую после острой фазы,но в пределах семи, шести, пяти или четырех дней после события, вызывающего рвоту). Рвота может составлять тошноту и рвоту, вызываемую химиотерапией ("CINV"), от умеренно эметогенной или в высшей степени эметогенной химиотерапии, тошноту и рвоту, вызываемую радиационной терапией("RTNV"), или послеоперационную тошноту и рвоту ("PONV"). Исследование биологической эквивалентности Когда о продукте говорят, что он демонстрирует конкретный фармакокинетический параметр "в пределах биологической эквивалентности", будет понятно, что продукт является биологически эквивалентным исследуемому лекарственному средству при использовании исследования биологической эквивалентности, описанного здесь. Исследование биологической эквивалентности, как правило, требует исследования in vivo на людях, у которых концентрация активного ингредиента или активного остатка и,когда нужно, его активного метаболита (метаболитов) в цельной крови, плазме, сыворотке или другой соответствующей биологической жидкости измеряется как функция времени. Определяемая как относительная биологическая доступность ("ВА"), биологическая эквивалентность ("BE") включает в себя сравнение между исследуемым и эталонным продуктом лекарственного средства. Хотя ВА и BE тесно связаны, сравнения BE обычно основываются на (1) критерии, (2) доверительном интервале для критерия и (3) на заданном пределе BE. Стандарт при конструировании исследований BE in vivo основывается на введении либо одной, либо множества доз исследуемого и эталонного продуктов здоровым субъектам в отдельных случаях, при случайном назначении двух возможных последовательностей введения лекарственного продукта. Статистический анализ фармакокинетических мер, таких как площадь под кривой (AUC) и пиковая концентрация (Cmax), предпочтительно основывается на так называемой "процедуре двухсторонних тестов", чтобы определить, являются ли средние значения фармакокинетических мер, определяемые после введения исследуемого и эталонного продуктов, сравнимыми. Этот подход называют средней биологической эквивалентностью, и он включает в себя вычисление 90% доверительного интервала для отношения средних значений (геометрических средних по популяции) для мер, для исследуемого и эталонного продуктов. Для установления BE, вычисленный доверительный интервал должен попасть в предел BE, то есть 80-125% для отношения средних значений для продукта. Таким образом, например, говорят, что биологическая эквивалентность установлена для данного набора обстоятельств с помощью 90% доверительного интервала для AUC, который находится в пределах между 80 и 125%, и 90% доверительного интервала для Cmax, который находится в пределах между 80 и 125%. Дополнительные детали относительно процедур BE можно найти в издаваемой FDA (администрации США по лекарственным препаратам и пищевым продуктам) инструкции, июль 1992 года, озаглавленной "Statistical Procedures for Bioequivalence Studies Using a Standard Two-Treatment Crossover Design," содержание которой включается сюда в качестве ссылки. Примеры Следующие далее примеры приводятся с тем, чтобы обеспечить специалистов в данной области полным объяснением и описанием того, как получают и оценивают соединения, заявленные здесь, и как предполагается, являются чистыми иллюстрациями настоящего изобретения и не предназначаются для ограничения рамок того, что авторы рассматривают как их изобретение. Делаются попытки обеспечить точность по отношению к числам (например, количеств, температуры, и тому подобное), но некоторые-9 016455 ошибки и отклонения должны приниматься во внимание. Если не утверждается иного, доли представляют собой массовые доли, температура приводится в С или находится при комнатной температуре, и давление является атмосферным или находится в пределах, близких к нему. Пример 1. Репрезентативное приготовление гелькапа. Таблица 1 описывает репрезентативные препараты твердой пероральной дозированной формы гелькапа, содержащей 0,25, 0,50 и 0,75 мг палоносетрона. Таблица 1. Репрезентативное приготовление гелькапа Пример 2. Протокол получения. Способ компаундирования включает в себя приготовление двух отдельных смесей, побочной смеси,содержащей активный ингредиент, глицерин и воду, и главной смеси, содержащей остальные наполнители. Способ начинают с двух отдельных смесей, которые затем объединяют, чтобы составить конечный раствор начинки для инкапсулирования. Раствор начинки выдерживают в атмосфере азота в течение фаз компаундирования и инкапсулирования. Пример 3. Протокол репрезентативного исследования растворения. Иллюстративный способ растворения для пероральных капсул с палоносетроном, 0,25 мг, 0,50 мг и 0,75 мг использует Устройство 2 USP (лопастная мешалка) при 75 об/мин в 500 мл 0,01 н. HCl при температуре растворения 37,00,5 С. Критерий приемлемости представляет собой "не менее чем 75% через 45 мин". Взвешивают индивидуально шесть мягких гелевых капсул. Мягкие гелевые капсулы помещают в каждую емкость, и отбирают образцы через 15, 30, 45 и 60 минут. Отбор через 15, 30, 60 минут осуществляют только для информации. Растворы образца извлекают и фильтруют через проточные фильтры в пробирки или флаконы для ВЭЖХ. Образцы анализируют с использованием системы ВЭЖХ с УФдетектором. Таблица 2. Условия растворения- 10016455 Пример 4. Химическая и физическая стабильность. Таблица 3 представляет результаты исследования химической и физической стабильности для 0,75 мг мягких гелевых препаратов палоносетрона, о которых сообщалось в примере 1, упакованных в блистерную упаковку 25 (формование: LM 15088, фольга: Reynolds 701). Таблица 3. Химическая и физическая стабильность Пример 5. Химическая и физическая стабильность. Таблица 4 представляет результаты исследования химической и физической стабильности для 0,50 мг мягких гелевых препаратов палоносетрона, о которых сообщалось в примере 1, упакованных в блистерную упаковку 25 (формование: LM 15088, фольга: Reynolds 701) . Таблица 4. Химическая и физическая стабильность Пример 6. Химическая и физическая стабильность. Таблица 5 представляет результаты исследования химической и физической стабильности для 0,25 мг мягких гелевых препаратов палоносетрона, о которых сообщалось в примере 1, упакованных в блистерную упаковку 25 (формование: LM 15088, фольга: Reynolds 701). Пример 7. Репрезентативный препарат для инъекций. Следующая далее таблица 6 описывает репрезентативный препарат для инъекций. Таблица 6. Репрезентативный препарат для инъекций Пример 8. Идентификация и анализ палоносетрона в мягких гелях с палоносетроном HCl с помощью ВЭЖХ с УФ-детектором. Процедура исследования Приготавливают растворы образца и стандарта при номинальной концентрации палоносетрон HCl 6,25 мкг/мл в 0,01 н. HCl. Фильтруют раствор и инжектируют его в систему ВЭЖХ. Условия ВЭЖХHCl и анализ в ходе способа раствора начинки мягких гелевых капсул. Процедура исследования Приготавливают растворы образца и стандарта при номинальной концентрации палоносетрона HCl 0,15 мг/мл в метаноле. Инжектируют растворы непосредственно в систему ВЭЖХ. Пример 10. Определение соединений, родственных палоносетрону, в мягких гелевых капсулах с палоносетроном HCl с помощью хиральной ВЭЖХ с УФ-детектором. Процедура исследования. Приготавливают раствор образца при номинальной концентрации палоносетрона HCl 0,34 мг/мл в метаноле. Приготавливают раствор стандарта Cpd2 при номинальной концентрации 5,6 мкг/мл. Приготавливают раствор для разрешения в метанольном растворителе, при номинальной концентрации 8 мкг/мл: Концентрация Cpd2, Cpd4, Cpd5, Cpd6 и Cpd7 приблизительно 0,4 мкг/мл. Инжектируют растворы непосредственно в систему ВЭЖХ. Условия ВЭЖХ Пример 11. Растворение мягких гелевых капсул палоносетрона HCl с помощью анализа ВЭЖХ. Процедура исследования Приготавливают растворы стандарта при номинальной концентрации палоносетрона HCl 1 мкг/мл вHCl 0,01 н. Раствор образца: помещают мягкую гелевую капсулу в емкость, содержащую 500 мл 0,01 н. HCl. Растворы фильтруют и инжектируют в систему ВЭЖХ. Условия ВЭЖХ Пример 12. Биологическая эквивалентность 0,75 мг гелькапа и дозированных форм для инъекций. Биологическая эквивалентность и абсолютная биологическая доступность исследуются для одной пероральной дозы двух препаратов по 0,75 мг палоносетрона на здоровых добровольцах. Исследование представляет собой исследование с тремя сеансами лечения, тремя периодами, с двумя перекрестными последовательностями. Лечение А представляет собой одну дозу из 0,75 мг палоносетрона в клиническом препарате гелькапа, описанном в таблице 1. Лечение В представляет собой одну дозу из 0,75 мг палоносетрона в коммерческом препарате гелькапа в таблице 1. Лечение IV состоит из трех последовательных инъекций болюса Aloxi, 25 мг. Фармакокинетические параметры приводятся ниже в табл. 7. Фармакокинетика приводится также на фиг. 1, где b1 представляет собой лечение препаратом А, b2 представляет собой лечение препаратом В и b3 представляет собой лечение Aloxi i.v., на фигуре изображены среднеарифметические концентрации палоносетрона в плазме (нг/мл) в зависимости от времени (ч) в линейном масштабе (n=33). Пример 13. Биологическая эквивалентность 50 мг клинических и коммерческих препаратов гелькапов. Исследования биологической эквивалентности предпринимают для оценки отдельных пероральных доз двух препаратов (препарата А и препарата В). палоносетрона в 0,50 мг мягких гелевых капсулах на здоровых субъектах мужского и женского пола. Исследование представляет собой два сеанса лечения,исследование с двумя периодами, двумя последовательностями, без плацебо, неупорядоченное перекрестное исследование. Фармакокинетические результаты приводятся в табл. 8. Фармакокинетические параметры также приводятся на фиг. 2, где N представляет собой клинический препарат А и b2 представляет собой коммерческий препарат В. На фигуре приведены среднеарифметические концентрации палоносетрона в плазме (нг/мл) в зависимости от времени (ч) в линейном масштабе (n=33). В настоящей заявке упоминаются различные публикации. Описание этих публикаций во всей их полноте тем самым включается в качестве ссылок в настоящую заявку, чтобы более полно описать состояние области, к которой относится настоящее изобретение. Специалистам в данной области будет понятно, что различные модификации и вариации могут быть проделаны в настоящем изобретением без отклонения от рамок или духа настоящего изобретения. Другие варианты осуществления настоящего изобретения будут ясны специалистам в данной области из рассмотрения описания и осуществления изобретения, описанного здесь. Предполагается, что описание и примеры должны рассматриваться только как иллюстрации, при этом истинные рамки и дух настоящего изобретения показаны с помощью следующей далее формулы изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Мягкая желатиновая капсула для перорального введения, содержащая:a) мягкую желатиновую наружную оболочку, имеющую проницаемость для кислорода, меньшую,чем примерно 1,010-3 млсм/(см 224 чатм); иb) липофильную жидкую композицию внутренней начинки, содержащую:i) больше чем примерно 50 мас.% одного или нескольких липофильных компонентов;(ii) от примерно 1 до примерно 20 мас.% воды, перемешанной или гомогенизированной в указанных одном или нескольких липофильных компонентах;(iii) от примерно 0,05 до примерно 2,0 мг палоносетрона гидрохлорида, солюбилизированного или диспергированного в указанной воде; иiv) поверхностно-активное вещество,где указанная капсула при пероральном приеме в голодном состоянии демонстрирует фармакокинетику, которая биологически эквивалентна препарату, имеющему более чем 95% абсолютную биологическую доступность, причем биологическая эквивалентность устанавливается с помощью 90% доверительного интервала для AUC, который находится в пределах между 80 и 125%.- 15016455 2. Капсула по п.1, в которой указанная композиция внутренней начинки содержит:a) от 0,5 до 1,0 мг палоносетрона гидрохлорида иb) солюбилизирующее эффективное количество жидкости, содержащей липофильный наполнитель и воду. 3. Мягкая желатиновая капсула по п.1, содержащая глицерин в указанной наружной оболочке и указанную композицию внутренней начинки. 4. Мягкая желатиновая капсула по п.1, в которой:a) указанная композиция внутренней начинки дополнительно содержит антиоксидант или восстанавливающий агент;b) указанный палоносетрон содержит меньше чем примерно 1 мас.% (3S)-3-[(3aS)-1-оксо 2,3,3 а,4,5,6-гексагидро-1H-бензо[de]изохинолин-2-ил]-1-азониабицикло[2.2.2]октан-1-олат. 5. Капсула по п.1, демонстрирующая фармакокинетику при пероральном приеме в голодном состоянии, которая является биологически эквивалентной препарату, имеющему более чем 95% абсолютную биологическую доступность и Cmax от 800 до 820 нг/л, где биологическая эквивалентность устанавливается с помощью:a) 90% доверительного интервала для AUC, который находится в пределах между 80 и 125%, иb) 90% доверительного интервала для Cmax, который находится в пределах между 80 и 125%. 6. Капсула по п.1, в которой указанная композиция внутренней начинки содержит кислород в количестве, которое опосредует не более чем примерно 3,0 мас.% окислительной деградации, когда указанная дозированная форма хранится в течение трех месяцев при 40 С и 75% RH. 7. Капсула по п.1, в которой не менее чем примерно 75% указанного палоносетрона или его фармацевтически приемлемой соли растворяется через 45 мин, когда исследуется в устройстве для растворения типа II с лопастной мешалкой в соответствии с Фармакопеей США при 75 об./мин и 37 С в 500 мл 0,01 н.HCl. 8. Капсула по п.1, в которой не менее чем примерно 75% указанного палоносетрона или его фармацевтически приемлемой соли растворяется через 30 мин, когда исследуется в устройстве для растворения типа II с лопастной мешалкой в соответствии с Фармакопеей США при 75 об./мин и 37 С в 500 мл 0,01 н.HCl. 9. Капсула по п.1, в которой указанная оболочка имеет проницаемость для кислорода, меньшую чем примерно 1,010-4 млсм/(см 224 чатм). 10. Капсула по п.1, в которой указанная внутренняя начинка содержит от 0,5 до 4 мас.% поверхностно-активного вещества. 11. Дозированная форма по п.1, в которой указанный палоносетрон или его фармацевтичеки приемлемая соль содержит (3S)-3-[(3aS)-1-оксо-2,3,3 а,4,5,6-гексагидро-1H-бензо[de]изохинолин-2-ил]-1 азониабицикло[2.2.2]октан-1-олат или ее фармацевтически приемлемую соль в количестве меньшем, чем 1,0 мас.% по отношению к массе указанного палоносетрона, и где указанная композиция внутренней начинки содержит кислород в количестве, которое опосредует не более чем примерно 3,0 мас.% окислительной деградации указанного палоносетрона или его фармацевтически приемлемой соли, когда указанная дозированная форма хранится три месяца или больше при 40 С и 75% RH. 12. Капсула по п.11, демонстрирующая фармакокинетику при пероральном приеме в голодном состоянии, которая является биологически эквивалентной препарату, имеющему более чем 90% абсолютную биологическую доступность, где биологическая эквивалентность устанавливается с помощью 90% доверительного интервала для AUC, который находится в пределах между 80 и 125%. 13. Капсула по п.11, в которой указанная композиция внутренней начинки содержит от примерно 1 мас.% до примерно 20 мас.% воды. 14. Капсула по п.11, демонстрирующая фармакокинетику при пероральном приеме в голодном состоянии, которая является биологически эквивалентной препарату, имеющему более чем 95% абсолютную биологическую доступность, и Cmax от 800 до 820 нг/л, где биологическая эквивалентность устанавливается с помощью:a) 90% доверительного интервала для AUC, который находится в пределах между 80 и 125%, иb) 90% доверительного интервала для Cmax, который находится в пределах между 80 и 125%. 15. Капсула по п.11, в которой не менее чем примерно 75% указанного палоносетрона или его фармацевтически приемлемой соли растворяется через 45 мин, когда исследуется в устройстве для растворения типа II с лопастной мешалкой в соответствии с Фармакопеей США при 75 об./мин и 37 С в 500 мл 0,01 н. HCl. 16. Капсула по п.11, в которой указанная оболочка имеет проницаемость для кислорода, меньшую чем примерно 1,010-4 млсм/(см 224 чатм). 17. Капсула по п.11, в которой:a) указанная композиция внутренней начинки содержит глицерин иb) указанная оболочка содержит глицерин. 18. Капсула по п.11, в которой указанная оболочка содержит желатин, целлюлозу, крахмал или- 16016455 НРМС. 19. Способ оптимизации биологической доступности и стабильности палоносетрона в желатиновой капсуле, содержащей палоносетрон по п.1, включающий в себя:a) создание мягкой желатиновой наружной оболочки, имеющей проницаемость для кислорода менее чем примерно 1,010-3 млсм/(см 224 чатм); иb) получение композиции начинки с помощью стадий, включающих в себя:i) получение от примерно 0,05 до примерно 2,0 мг палоносетрона в виде палоносетрона гидрохлорида, где указанный палоносетрон содержит (3S)-3-[(3aS)-1-оксо-2,3,3 а,4,5,6-гексагидро-1H-бензоii) растворение или диспергирование указанного палоносетрона в воде с образованием водного премикса;iii) смешивание указанного водного премикса с одним или несколькими липофильными наполнителями при массовом отношении водного премикса к липофильным наполнителям меньше чем 30:70, с образованием перемешанной или гомогенной композиции липофильной начинки;iv) смешивание поверхностно-активного вещества с указанной водой, указанным водным премиксом или указанной композицией начинки; иv) балансирование количеств поверхностно-активного вещества и воды в указанной композиции начинки для облегчения биологической доступности палоносетрона из указанной желатиновой капсулы при пероральном приеме и для сведения к минимуму степени деградации палоносетрона; иc) заполнение указанной наружной оболочки указанной композицией начинки. 20. Способ по п.19, в котором указанная композиция начинки содержит от примерно 0,1 до примерно 10,0 мас.% поверхностно-активного вещества и от примерно 0,1 до примерно 20 мас.% воды. 21. Способ по п.19, в котором указанная композиция начинки содержит от примерно 0,5 до примерно 4 мас.% поверхностно-активного вещества и от примерно 1 до примерно 10 мас.% воды. 22. Способ по п.19, в котором указанная наружная оболочка капсулы дополнительно содержит глицерин, дополнительно включающий в себя смешивание указанного водного премикса с глицерином до или после образования указанной композиции липофильной начинки. 23. Способ получения набора дозированных форм палоносетрона, имеющих пониженное количество примесей и продуктов деградации, опосредуемых кислородом, включающий в себя:a) смешивание палоносетрона гидрохлорида и одного или нескольких фармацевтически приемлемых наполнителей с образованием смеси по п.1;b) переработку указанной смеси в множество конечных дозированных форм по п.1; иc) исследование одной или нескольких указанных конечных дозированных форм на одно или несколько соединений, родственных палоносетрону, выбранных из (3S)-3-[(3aS)-1-оксо-2,3,3 а,4,5,6 гексагидро-1H-бензо[de]изохинолин-2-ил]-1-азониабицикло[2.2.2]октан-1-олата,2-[(3S)-1-азобицикло[2.2.2]окт-3-ил]-2,4,5,6-тетрагидро-2H-бензо[de]изохинолин-1-он гидрохлорида и (3aR)-2-[(S)-1-азобицикло[2.2.2]окт-3-ил]-2,3,3 а,4,5,6-гексагидро-1-оксо-1H-бенз[de]изохинолин гидрохлорида или их гидрохлоридной соли. 24. Способ по п.23, включающий в себя исследование на (3S)-3-[(3aS)-1-оксо-2,3,3 а,4,5,6 гексагидро-1H-бензо[de]изохинолин-2-ил]-1-азониабицикло[2.2.2]октан-1-олат или его гидрохлоридную соль. 25. Способ по п.23, включающий в себя исследование на 2-[(3S)-1-азобицикло[2.2.2]окт-3-ил]2,4,5,6-тетрагидро-1H-бензо[de]изохинолин-1-он гидрохлорида или его гидрохлоридную соль. 26. Способ по п.23, включающий в себя исследование на (3aR)-2-[(S)-1-азобицикло[2.2.2]окт-3-ил]2,3,3 а,4,5,6-гексагидро-1-оксо-1H-бенз[de]изохинолин гидрохлорида или его гидрохлоридную соль. 27. Способ по п.23, дополнительно включающий в себя исследование указанного палоносетрона гидрохлорида или указанной конечной дозированной формы на одно или несколько соединений, выбранных из (3S)-3-(1-оксо-2,4,5,6-тетрагидро-2H-бензо[de]изохинолин-2-ил)-1-азониабицикло[2.2.2]октан-1-олата, (3aR)-2-[(R)-1-азобицикло-[2.2.2]окт-3 ил]-2,3,3 а,4,5,6-гексагидро-1-оксо-2H-бенз[de]изохинолин гидрохлорида, (3aS)-2-[(R)-1-азобицикло[2.2.2]окт-3 ил]-2,3,3 а,4,5,6-гексагидро-1-оксо-2H-бенз[de] изохинолин гидрохлорида или (3aS)-2-[(S)-1-азобицикло[2.2.2]окт-3 ил]-2,3,3 а,4,5,6-гексагидро-1-оксо 2H-бенз[de]изохинолин гидрохлорида или их гидрохлоридной соли.
МПК / Метки
МПК: A61K 9/48, A61K 31/473, A61K 9/66
Метки: доступность, биологическую, стабильность, содержащие, палоносетрона, имеющие, капсулы, гидрохлорид, мягкие, улучшенную
Код ссылки
<a href="https://eas.patents.su/19-16455-myagkie-kapsuly-soderzhashhie-palonosetrona-gidrohlorid-imeyushhie-uluchshennuyu-stabilnost-i-biologicheskuyu-dostupnost.html" rel="bookmark" title="База патентов Евразийского Союза">Мягкие капсулы, содержащие палоносетрона гидрохлорид, имеющие улучшенную стабильность и биологическую доступность</a>
Мягкие композиции в форме куска, содержащие смеси полиалкиленгликолей

Номер патента: 1464
Опубликовано: 23.04.2001
Авторы: Петко Майкл Франсис, Пост Альберт Джозеф, Осмер Фредерик Сильвио
МПК: C11D 3/37
Метки: куска, форме, содержащие, полиалкиленгликолей, мягкие, смеси, композиции
Формула / Реферат:
1. Моющая композиция, содержащая: (а) 10-60 мас.% синтетического, не являющегося мылом моющего вещества или смеси синтетических, не являющихся мылом моющих веществ; (в) более 10%, но не более 60 мас.% водорастворимого полиалкиленгликольного детергента, который имеет точку плавления в интервале 40-100ш С; (c) 0,01-10 мас.% полиалкиленгликоля или смеси полиалкиленгликолей, которые (полиалкиленгликоль или смесь полиалкиленгликолей) имеют точку...
Жидкие фармацевтические композиции палоносетрона

Номер патента: 14066
Опубликовано: 30.08.2010
Авторы: Бонадео Даниеле, Канелла Роберта, Кальдерари Джорджо, Бралья Риккардо, Бралья Энрико
МПК: A61K 9/08, A61K 31/4747
Метки: композиции, палоносетрона, фармацевтические, жидкие
Формула / Реферат:
1. Фармацевтически стабильный раствор для профилактики или уменьшения рвоты, содержащий:a) от 0,03 до 0,2 мг/мл палоносетрона гидрохлорида;b) фармацевтически приемлемый носитель при рН от 4,0 до 6,0 и хелатирующий агент.2. Раствор по п.1, где концентрация палоносетрона гидрохлорида составляет 0,05 мг/мл.3. Раствор по п.1, где рН составляет от 4,5 до 5,5.4. Раствор по п.1, содержащий в качестве хелатирующего агента EDTA в концентрации от 0,005 до...
Гранулы, содержащие гидрохлорид венлафаксина

Номер патента: 9695
Опубликовано: 28.02.2008
Авторы: Фекете Паль, Мориц Эстер, Корбей Тибор, Божо Агнеш
МПК: A61K 47/02, A61K 47/38, A61K 31/137...
Метки: содержащие, гидрохлорид, гранулы, венлафаксина
Формула / Реферат:
1. Гранулы формы, близкой к сферической, включающие фармацевтически активный гидрохлорид венлафаксина, где указанные гранулы содержат (относительно общей массы), максимум, 80 мас.% гидрохлорида венлафаксина, от 10 до 60 мас.% хлорида натрия и/или хлорида калия, от 10 до 60 мас.% микрокристаллической целлюлозы и, возможно, другие фармацевтически приемлемые эксципиенты и/или добавки, способствующие грануляции. 2. Гранулы по п.1, имеющие размер...
Обойма капсулы и способ извлечения капсулы из обоймы

Номер патента: 3801
Опубликовано: 30.10.2003
Автор: Коллеп Александр
МПК: A47J 31/40
Метки: извлечения, обойма, способ, капсулы, обоймы
Формула / Реферат:
1. Обойма капсулы, выполненная с возможностью её установки в кофейном автомате, содержащая канал поступления воды и корпус для извлекаемой капсулы, отличающаяся тем, что содержит непосредственно на выходе канала поступления воды и в корпусе систему, пробивающую капсулу и автономную от канала поступления воды и имеющую, по меньшей мере, одно лезвие; при этом лезвие содержит две боковые кромки и режущую кромку, причём одна кромка из числа двух...
Имеющие противоопухолевую активность комплексы платины, содержащие химически модифицированные желчные кислоты

Номер патента: 8221
Опубликовано: 27.04.2007
Авторы: Бартоли Энцо, Медичи Алессандро, Палмиери Бениамино
МПК: A61K 31/58, A61P 35/00, C07J 51/00...
Метки: содержащие, кислоты, комплексы, активность, имеющие, противоопухолевую, платины, модифицированные, желчные, химически
Формула / Реферат:
1. Комплекс платины(II) формулы в которой n равно 0 или 1 и А представляет собой С или группу СН-СН. 2. Комплекс по п.1, где n равно 0 и А представляет собой группу СН-СН. 3. Комплекс по п.1, где n равно 1 и А представляет собой С. 4. Применение комплекса по п.1 в качестве лекарственного средства, обладающего противоопухолевой активностью. 5. Фармацевтическая композиция, имеющая противоопухолевую активность и содержащая комплекс по любому из...
Предыдущий патент: Способ усиления сосудов неправильной формы, работающих под давлением
Следующий патент: 8-сульфонил-1,3,4,8-тетрагидро-2н-[1,4]оксазепино[6,7-e]индольные производные и их использование в качестве 5-нт6 рецепторных лигандов
Случайный патент: Способ получения полипептида или вируса, представляющих интерес, в непрерывной клеточной культуре