Способ получения 2-(замещенный фенил)-2-гидроксиэтилкарбаматов












Формула / Реферат
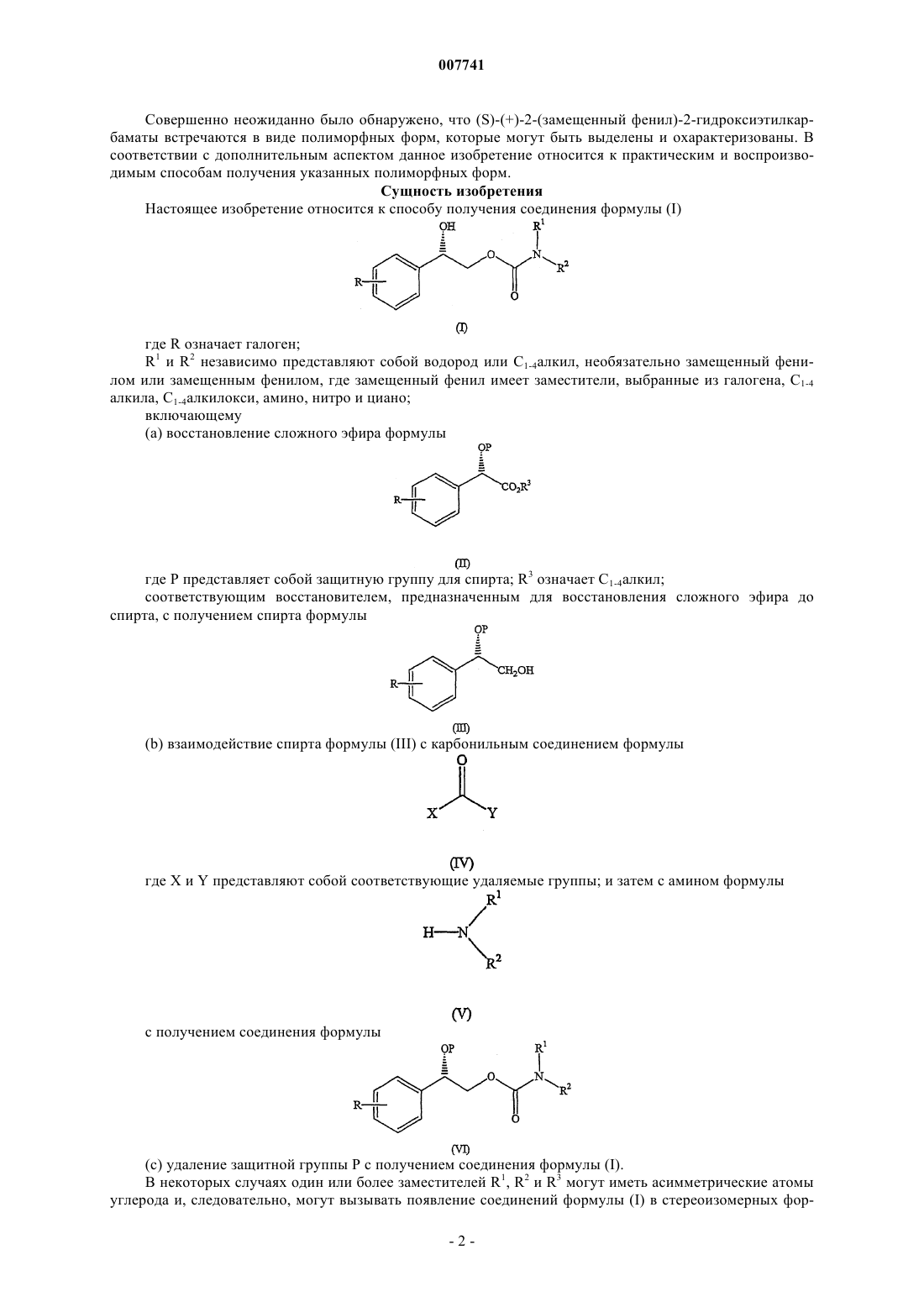
1. Способ получения соединения формулы

где R означает галоген;
R1 и R2 независимо представляют собой водород или С1-4алкил,
включающий
(а) восстановление сложного эфира формулы

где Р представляет собой соответствующую защитную группу для спирта;
R3 означает С1-4алкил;
соответствующим восстановителем сложного эфира до спирта, с получением спирта формулы

(b) взаимодействие спирта формулы (III) с карбонильным соединением формулы

где X и Y представляют собой соответствующие удаляемые группы; и затем с амином формулы

с получением таким образом соединения формулы

(с) удаление защитной группы Р с получением соединения формулы I.
2. Способ по п.1, в котором R представляет собой 2-хлор, R1 и R2 представляют собой водород.
3. Способ по п.1 или 2, в котором защитная группа для спирта выбрана из 2-(2-метокси)пропила.
4. Способ по п.1 или 2, в котором карбонильное соединение формулы (IV) выбрано из диимидазолилкарбонила, арил- или замещенный арилгалогенформиата.
5. Способ по п.1 или 2, в котором R3 означает метил.
6. Способ по п.1 или 2, в котором соответствующий восстановитель сложного эфира до спирта представляет собой гидрид металла или комплекс гидрида металла.
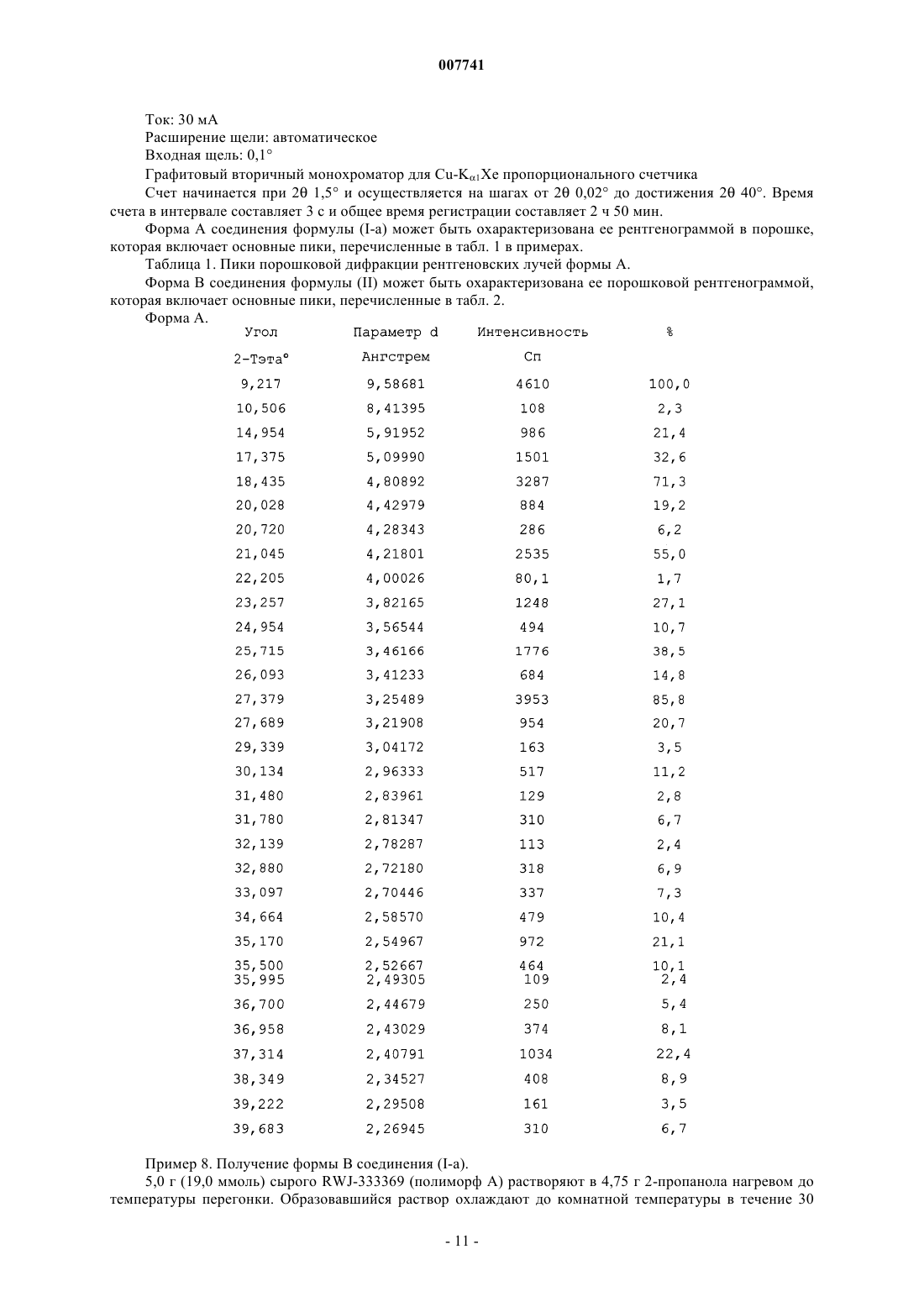
7. Соединение формулы

где R, R1 и R2 являются такими, как определено в п.1 или 2, и Р является группой типа простого эфира.
8. Соединение по п.7, где группа типа простого эфира выбрана из метоксиметиловых эфиров (MOM);
тетрагидропирановых эфиров (ТНР эфиров);
хлорзамещенных тетрагидрофураниловых эфиров, полученных из 2-хлортетрагидрофурана;
тетрагидротиофураниловых эфиров;
1-этоксиэтиловых эфиров;
1-метил-1-метоксиэтиловых эфиров;
трифениловых эфиров и их соответствующих производных;
бензиловых эфиров и их соответствующих производных;
4-метокситетрагидропираниловых эфиров;
4-метокситетрагидротиопираниловых эфиров.
9. Соединение по любому из пп.7 или 8, где Р представляет собой 2-(2-метокси)пропил.
10. Соединение формулы (I-а)

отличающееся тем, что соединение формулы (I-а) находится в моноклинной кристаллической форме.
11. Соединение формулы (I-а), как определено в п.10, имеющее следующие пики дифракции рентгеновских лучей в порошке

12. Соединение по п.10, по существу, не содержащее другие полиморфные формы соединения формулы (I-а).
13. Соединение по п.11, по существу, не содержащее другие полиморфные формы соединения формулы (I-а).
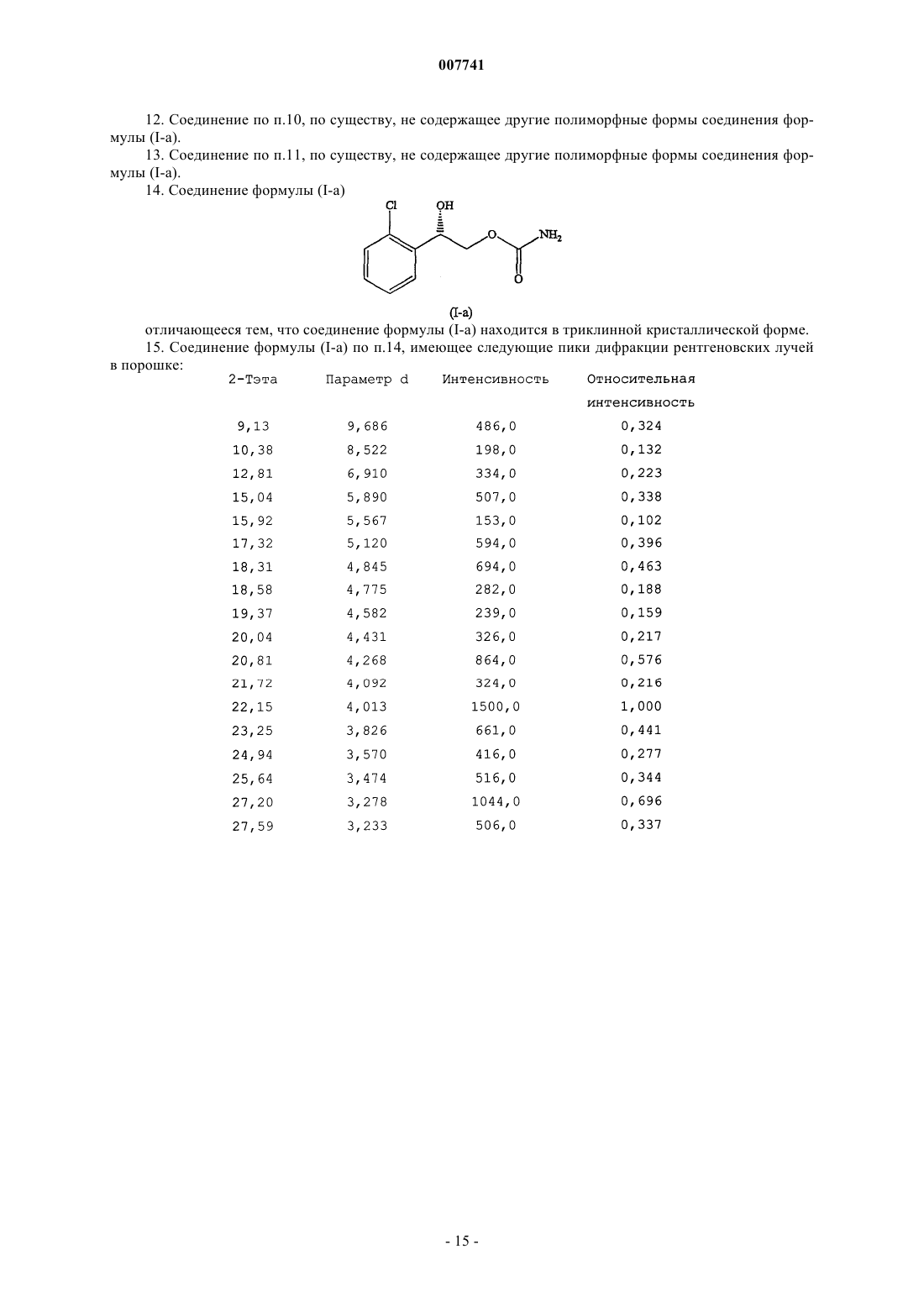
14. Соединение формулы (I-а)

отличающееся тем, что соединение формулы (I-а) находится в триклинной кристаллической форме.
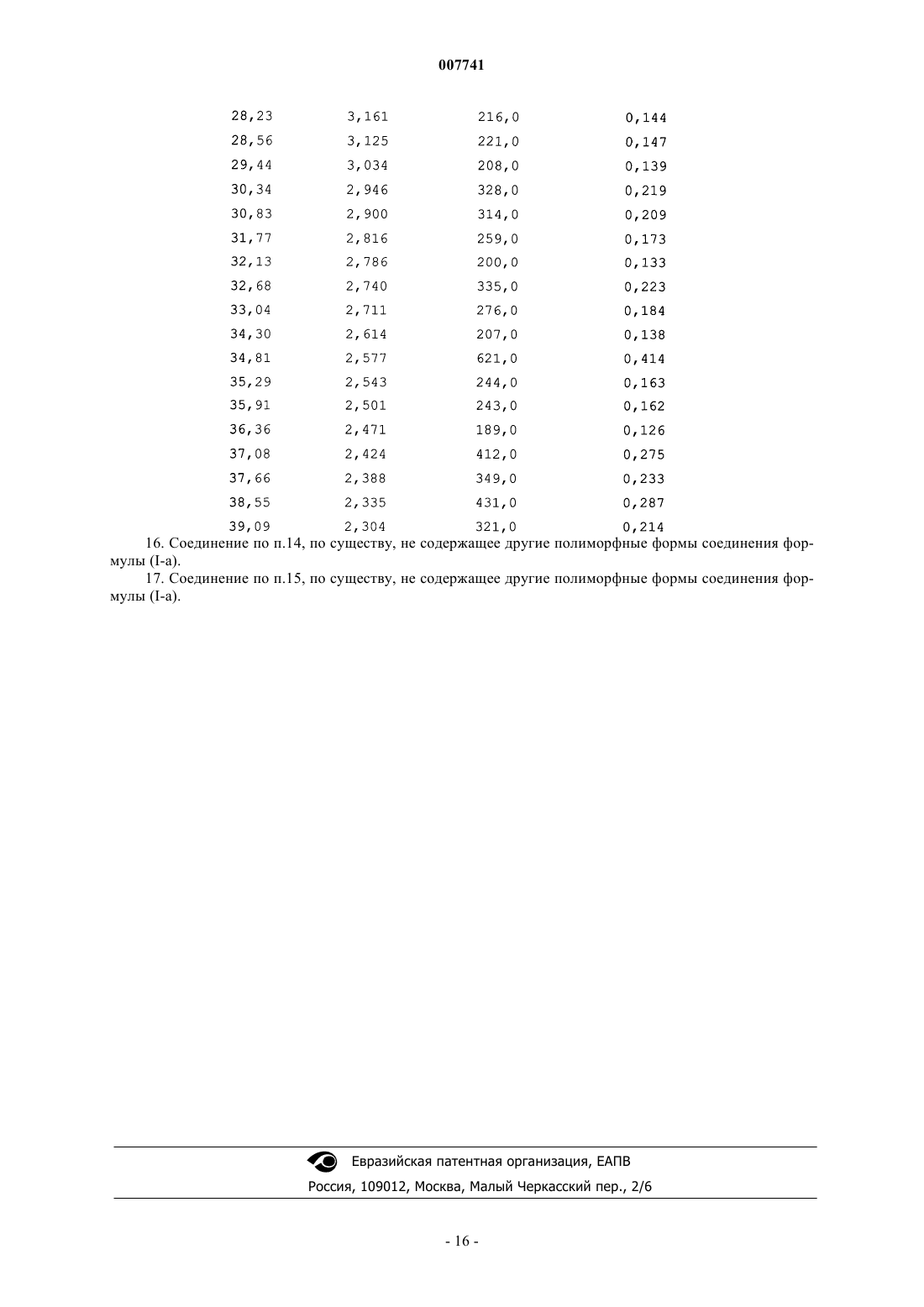
15. Соединение формулы (I-а) по п.14, имеющее следующие пики дифракции рентгеновских лучей в порошке:


16. Соединение по п.14, по существу, не содержащее другие полиморфные формы соединения формулы (I-а).
17. Соединение по п.15, по существу, не содержащее другие полиморфные формы соединения формулы (I-а).
Текст
007741 Краткое описание изобретения Настоящее изобретение относится к новому способу получения (S)-(+)-2-(замещенный фенил)-2 гидроксиэтилкарбаматов и к новым промежуточным продуктам, использованным в данном способе. Кроме того, оно относится к полиморфным формам указанных карбаматов. Предшествующий уровень изобретения 2-(Замещенный фенил)-2-гидроксиэтилкарбаматы и их изомеры раскрыты в патенте США 5698588 и патенте США 5854283, который соответствует WO 97/26241, в качестве соединений, применимых для лечения расстройств центральной нервной системы, в частности судорог, эпилепсии, удара, мышечных спазм, невропатической боли и мигрени. Указанные соединения имеют асимметрический атом углерода в бензильном положении, который представляет собой алифатический атом углерода, смежный с фенильным кольцом. Асимметрический атом углерода предпочтительно находится в S-конфигурации. Предпочтительное соединение среди указанных карбаматов представляет собой (S)-(+)-2-(2-хлорфенил)2-гидроксиэтилкарбамат, при этом данное соединение обозначают как RWJ-333369. Указанная форма и подобные оптически чистые формы карбаматов галогенированного 2-фенил-1,2-этандиола раскрыты в патенте США 6127412 и в патенте США 6103759. Указанные соединения получают взаимодействием соответствующим образом замещенного фенил 1,2-этандиола с диметилкарбонатом, при этом образуется соответствующий фенил-1,2-этандиолкарбонат, который затем подвергают взаимодействию с подходящим амином, который дает две региоизомерные формы образовавшихся карбаматов, которые должны быть разделены колоночной хроматографией. Подобным образом получали стереоизомерно чистые соединения, исходя из энантиомерно чистых исходных соединений. В результате осуществления указанного способа получают смесь двух региоизомерных продуктов, один из которых необходимо удалить, поскольку он оказывает отрицательное влияние на выход. Оба региоизомерных конечных продукта разделяют хроматографией, которая является обременительной процедурой, в частности, в крупномасштабном производстве. Задачей настоящего изобретения является обеспечение способа, который дает возможность не только избежать указанной стадии разделения и сопутствующей потери нежелательного региоизомера,но также получить конечный продукт с высоким выходом и высокой степенью чистоты, в частности, с высокой степенью энантиомерной чистоты. Другой объект изобретения заключается в создании способа,который может быть использован в производственном масштабе, и в котором используются исходные продукты, которые легко получить или которые являются коммерчески доступными. Еще одна задача состоит в создании способа, который является экономически эффективным и не вызывает образование побочных продуктов, которые являются опасными или трудноудаляемыми. Дополнительная задача настоящего изобретения состоит в создании способа, который имеет ограниченное число стадий очистки, в частности, способа, в котором нет необходимости в очистке некоторых или всех промежуточных продуктов. Еще одной задачей являлась разработка способа, который позволил бы избежать использования избытков исходного продукта и растворителей. Указанные задачи достигаются реализацией способа настоящего изобретения. Лекарственные вещества, отнесенные также к активным фармацевтическим ингредиентам, могут встречаться в виде полиморфных форм. Почти во всех случаях конкретная полиморфная форма имеет другие свойства по сравнению со свойствами активного ингредиента, не находящегося в специфической полиморфной форме, или по сравнению с другой полиморфной формой активного ингредиента. Могут отличаться свойства, касающиеся растворимости, стабильности, текучести, способности распадаться в пищеварительном тракте, прессуемости и т.д. Известно, что такие различия в указанных свойствах оказывают неблагоприятное действие на свойства составов, полученных из таких активных ингредиентов, а также изготовленных из них конечных или завершенных лекарственных форм. Во многих странах организации, ответственные за одобрение лекарственных средств, требуют полной характеристики активного ингредиента, использованного в каждом лекарственном препарате, включая идентификацию и контроль полиморфных форм. В случае присутствия их в конечном продукте, органы, утверждающие лекарственный препарат, требуют от производителя активного лекарственного вещества, по меньшей мере, осуществления контроля процессов синтеза с тем, чтобы содержание в процентах различных соответственных полиморфных форм, в случае их присутствия, соответствовали загрузкам и спецификациям, утвержденным для лекарственных препаратов. В результате отсутствия контроля процентное содержание данного полиморфа в процессах синтеза может изменяться, вследствие чего оказывается неблагоприятное влияние на свойства активного вещества и конечного лекарственного препарата, который теперь уже не будет удовлетворять спецификациям, в соответствии с которыми утверждается лекарственное средство. Поэтому необходим строгий контроль условий процесса синтеза и конечного продукта, в частности, что касается присутствия допустимых количеств полиморфных форм. Многие активные вещества не показывают полиморфизм и присутствие полиморфных форм нового химического ингредиента является не всегда предсказуемо. Изменение параметров процесса может привести к изменению степени присутствия полиморфных форм в конечных продуктах, которое, как объяснялось выше, является нежелательным. Поэтому получение информации о присутствии полиморфных форм и их характеристик является весьма желательной целью изобретения.-1 007741 Совершенно неожиданно было обнаружено, что (S)-(+)-2-(замещенный фенил)-2-гидроксиэтилкарбаматы встречаются в виде полиморфных форм, которые могут быть выделены и охарактеризованы. В соответствии с дополнительным аспектом данное изобретение относится к практическим и воспроизводимым способам получения указанных полиморфных форм. Сущность изобретения Настоящее изобретение относится к способу получения соединения формулы (I)R1 и R2 независимо представляют собой водород или С 1-4 алкил, необязательно замещенный фенилом или замещенным фенилом, где замещенный фенил имеет заместители, выбранные из галогена, С 1-4 алкила, С 1-4 алкилокси, амино, нитро и циано; включающему(а) восстановление сложного эфира формулы где Р представляет собой защитную группу для спирта; R3 означает С 1-4 алкил; соответствующим восстановителем, предназначенным для восстановления сложного эфира до спирта, с получением спирта формулы(b) взаимодействие спирта формулы (III) с карбонильным соединением формулы где X и Y представляют собой соответствующие удаляемые группы; и затем с амином формулы(с) удаление защитной группы Р с получением соединения формулы (I). В некоторых случаях один или более заместителей R1, R2 и R3 могут иметь асимметрические атомы углерода и, следовательно, могут вызывать появление соединений формулы (I) в стереоизомерных фор-2 007741 мах. Такие стереоизомерные формы предназначены для включения в объем настоящего изобретения. Предпочтительные соединения формулы (I) представляют такие соединения, где R означает 2-хлор,R1 и R2 представляют собой водород. Группа Р является соответствующей защитной группой для спирта. Предпочтительные группы Р представляют собой группы типа простого эфира. В частности, предпочтительной защитной группой Р является 2-(2-метокси)пропил. В предпочтительном варианте осуществления способа карбонильное соединение формулы (IV) выбрано из 1,1'-карбонилдиимидазола и фенилхлорформиата. В конкретном варианте осуществления R3 означает метил. Другое осуществление способа является таким, в котором соответствующий восстановитель сложного эфира в спирт представляет собой гидрид металла или комплексный гидрид металла. В соответствии с другим аспектом изобретение относится к соединению формулы где R, R1 и R2 являются такими, как определено в п.1 или 2 формулы изобретения, и Р является соответствующей защитной группой для гидрокси. В соответствии с другим аспектом изобретение относится к соединению формулы где R является таким, как определено в п.1 или 2 формулы изобретения, и Р является соответствующей защитной группой для гидрокси. В соответствии с еще одним другим аспектом изобретение относится к соединению формулы где R является таким, как определено в п.1 или 2 формулы изобретения, R3 означает С 1-4 алкил; и Р является соответствующей защитной группой для гидрокси. Предпочтительны соединения формул (IV), (III) или (II), где Р представляет собой 2-(2-метокси)пропил. Предпочтительны также соединения формулы (II), где R3 означает метил. В соответствии еще c одним аспектом изобретение относится к способу получения соединения формулы (VI), характеризующемуся указанными выше стадиями способа (а) и (b). В соответствии еще c одним аспектом изобретение относится к способу получения соединения формулы (III), характеризующемуся указанной выше стадией способа (а). В соответствии с еще одним аспектом изобретение относится к способу получения соединения формулы (II), характеризующемуся указанными выше стадиями способа (d) и (е). Изобретение дополнительно относится к способу получения соединения формулы (I), характеризующемуся стадиями способа (d) и (е), которые будут раскрыты далее, и стадиями способа (а), (b) и (с),указанными выше. В соответствии с дополнительным аспектом данное изобретение касается присутствия полиморфных форм (S)-(+)-2-(замещенный фенил)-2-гидроксиэтилкарбаматов. В частности, оно касается двух полиморфных форм соединения 2-(2-хлорфенил)-2-(2-(2-метокси)пропил)этилкарбамат. Изобретение также относится к способам получения указанных новых полиморфных форм. Подробное описание изобретения Предметом настоящего изобретения является способ получения указанных выше соединений формулы (I) и промежуточных продуктов формулы (II), (III), (IV) и (V), представленных и определенных выше. Предпочтительны соединения и промежуточные продукты, определенные в данном описании, где R представляет собой 2-хлор, R1 и R2 представляют собой водород. Соединение формулы (I), где заместители имеют последние указанные значения, может быть названо как 'RWJ-333369' и представлено струк-3 007741 турной формулой Термин галоген относится к фтору, хлору, брому и йоду. С 1-4 алкил означает насыщенные углеводородные радикалы с прямой и разветвленной цепью,имеющие от 1 до 4 атомов углерода, такие как метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2 метил-1-пропил, 2-метил-2-пропил. С 1-4 алкилокси означает С 1-4 алкильные радикалы, связанные с атомом кислорода, такие как метокси, этокси, 1-пропокси, 2-пропокси, 1-бутокси, 2-бутокси и подобные. Замещенный фенил представляет собой фенил, замещенный вышеуказанными заместителями. Замещенный фенил предпочтительно имеет 1, 2 или 3 заместителя. Предпочтительный заместитель представляет собой галоген, более предпочтительно хлор. Р, как указывалось выше, является соответствующей защитной группой для гидрокси. Предпочтительно ее выбирать таким образом, чтобы она была устойчивой в ходе стадии восстановления (II) в (III),а также на последующей стадии реакции превращения (III) в (VI). Предпочтительная операция восстановления включает использование комплексных гидридов, которые будут указаны ниже, при этом группа Р должна быть устойчивой по отношению к указанным комплексным гидридам и к полученным из них продуктам реакции. Для восстановления комплексными гидридами требуются щелочные условия и поэтому группа Р не должна расщепляться в щелочной среде. Группа Р должна быть предпочтительно способна удаляться в кислой среде, которая должна быть такой, чтобы карбаматная функциональная группа не расщеплялась. В частности, предпочтительны защитные группы Р, которые способны удаляться при рН, равном около 1 или чуть выше. В частности, применимы группы Р типа простого эфира. В качестве примеров группы Р могут быть указаны метоксиметиловые эфиры (MOM); которые могут быть получены из метоксиметилхлорида или формальдегиддиметилацеталя; тетрагидропирановые эфиры (ТНР эфиры), полученные из дигидропирана; тетрагидротиопираниловые эфиры, полученные из дигидротиопирана; хлорзамещенные тетрагидрофураниловые эфиры, полученные из 2-хлортетрагидрофурана; тетрагидротиофураниловые эфиры, полученные из дигидротиофурана; 1-этоксиэтиловые эфиры, полученные из этилвиниловых эфиров или 1-этоксиэтилхлорида; 1-метил-1-метоксиэтиловые эфиры, полученные из метилвиниловых эфиров, которые представляют особый интерес; трифениловые эфиры и их соответствующие производные, которые могут быть получены из соответствующих хлоридов; бензиловые эфиры и их соответствующие производные, которые могут быть получены из соответствующих бромидов или йодидов; 4-метокситетрагидропираниловые эфиры, полученные из 5,6-дигидро-4-метокси-2 Н-пирана; 4-метокситетрагидротиопираниловые эфиры, полученные из 5,6-дигидро-4-метокси-2 Н-тиопирана. Еще более выгодно использовать такие группы Р, которые не приводят к получению смеси диастереоизомеров, т.е. группы Р без асимметрического центра. При определенных обстоятельствах, например,в зависимости от природы восстановителя, в качестве соответствующих групп Р могут быть использованы отдельные силиловые эфиры, в частности, трет-бутилдиметилсилиловый (TBDMS), триизопропилсилиловый, трибензилсилиловый и подобные. Стадия (а). Получение 2-(замещенный фенил)-2-(защищенный гидрокси)этанола (III). В соответствии с первой стадией способа настоящего изобретения сложный эфир (II) восстанавливают до соответствующего спирта формулы (III) с использованием соответствующего восстановителя,предназначенного для восстановления сложного эфира в спирт. Восстановитель может представлять собой гидрид металла или комплексный гидрид металла, такой как алюмогидрид лития, или его производные. Особые восстановители для указанной реакции представляют собой силановые агенты, такие как триалкилсиланы, диалкилсиланы, триалкоксисиланы и предпочтительно полиметилводородсилоксан('PMHS'), в присутствии подходящего катализатора. Последний, в частности, представляет собой галогенид или карбоксилат переходного металла и предпочтительно последним является карбоксилат цинка,такой как гексаноат цинка или его производное, более предпочтительно 2-этилгексаноат цинка, в присутствии гидрида металла, такого как гидрид щелочного металла или щелочно-земельного металла или гидрид алюминия, например, гидрида лития, натрия, калия, кальция, или комплексного гидрида, такого как боргидрид или алюмогидрид, в частности боргидрид или алюмогидрид щелочного металла, напри-4 007741 мер, в присутствии боргидрида или алюмогидрида лития, натрия или калия. В качестве каталитической смеси наиболее предпочтительно используется комбинация, состоящая из 2-этилгексаноата цинка и боргидрида натрия. Указанные и подобные восстановители представлены в заявке на патент WO 96/12694(1995) и в: J. Ulman, The Alembic, 1999, 59, 1 ff. Реакцию данной стадии способа проводят в подходящем растворителе, например, простом эфире или простом полиэфире или углеводороде, в частности в ароматическом углеводороде. Конкретные примеры подходящих растворителей включают диэтиловый эфир, дипропиловый эфир, диизопропиловый эфир, дибутиловый эфир, диглим и толуол. Восстановитель может быть образован перемешиванием смеси реагентов ERS А и ERS В в отдельном реакционном сосуде (ERS = система восстановления сложного эфира, коммерчески доступная от RohmHaas). ERS А представляет собой раствор NaBH4 в тетраглиме, тогда как ERS В представляет собой раствор Zn (карбоксилат) 2.Н 2O, в частности, Zn (2-этилгексаноат)2 Н 2 О в тетрагидрофуране. ERS А и ERS В предпочтительно смешивают при повышенной температуре, например, при температуре в диапазоне от 50 до 90 С, в частности при 70 С, в течение нескольких минут, например в течение 30 мин. Затем смесьERS А и ERS В добавляют к сложному эфиру (II) и добавляют ERS С. ERS С представляет собой полиметилводородсилоксан. Восстановитель может быть также получен in situ в реакционном сосуде, что является особенно привлекательным. В таком варианте промежуточный продукт (II) растворяют в подходящем растворителе, предпочтительно в толуоле, при повышенной температуре 80-90 С. Сначала одной порцией добавляют ERS В, затем также одной порцией добавляют ERS А. Затем сразу же в течение одного часа добавляют ERS С с одновременным регулированием температуры и поддержанием ее при повышенных значениях, в частности, примерно при 95 С. Исходный сложный эфир миндальной кислоты (II) получают как указано ниже и он может быть выделен и необязательно очищен и использован как таковой на стадии восстановления. Сложный эфир(II) может также храниться в растворенном виде в растворителе, в котором он получен, и использоваться как таковой на стадии восстановления. Сложный эфир замещенной миндальной кислоты (II) предпочтительно представляет собой сложный метиловый эфир. В частном варианте осуществления указанной стадии способа исходный сложный эфир (II) или используют растворенным в растворителе предшествующей реакционной стадии или он может быть растворен в подходящем растворителе, например таком, как простой эфир, например в простом ди-нбутиловом эфире или предпочтительно в толуоле. Полученный, как указывалось выше, восстановитель добавляют одной порцией к раствору, затем в течение одного часа добавляют ERS С. Реакционная смесь перед добавлением ERS С может быть необязательно нагрета до 90 С. Затем температуру повышают до 90 С и поддерживают при указанной температуре до тех пор, пока при контроле способа степень конверсии будет составлять по меньшей мере 99%. Время протекания реакции составляет примерно 1 ч. После завершения восстановления смесь охлаждают до температуры 15-20 С и сначала осторожно гидролизуют метанолом и затем водным раствором гидроксида щелочного металла, которым предпочтительно является раствор NaOH, взятый в небольшом избытке (например, 1,3 мол.экв. при использовании 30% раствора). Затем смесь кипятят с обратным холодильником в течение 1 ч при температуре около 50 С и при комнатной температуре разделяют слои. Органический слой промывают водой и насыщенным водным раствором NaCl при комнатной температуре. Количество ERS С находится в диапазоне от 2 до 4, в частности от 2,2 до 3 мол.экв., предпочтительно используют 2,3 мол.экв. ERS С. Реакция обычно завершается после ее протекания в течение примерно 1 ч. После завершения реакции восстановления избыток ERS С может быть разрушен подходящим сложным эфиром, в частности этилацетатом. После добавления указанного сложного эфира смесь перемешивают в течение одного часа и гидролизуют при 90 С добавлением водного раствора основания,предпочтительно раствора NaOH или KОН (например, 33%), и затем предпочтительно с использованием метанола. Дальнейшая обработка включает отделение органического слоя и промывку водным щелочным раствором (например, 33% раствором KОН) и водой. Образовавшийся сложный метиловый эфир представляет собой масло и, в случае необходимости,может быть подвергнут перегонке для очистки. Стадия (b). Получение 2-(замещенный фенил)-2-(защищенный гидрокси) этил карбамата (VI). На данной стадии спирт формулы (III) подвергают взаимодействию с карбонильным соединением формулы где X и Y представляют собой соответствующие удаляемые группы. Предпочтительно одна из группы X или Y является более химически активной, чем другая. X и Y могут представлять собой галоген, в частности хлор или бром, но предпочтительно X и Y представляют собой имидазолильные группы. Если X является галогеном, Y предпочтительно представляет собой арилокси или алкоксильную группу. В последнем случае (V) является алкил- или арилгалогенформиатом. Конкретные арильные группы в(IV) представляют собой фенил или замещенный фенил, например галогенфенил, или С 1-4 алкил. Предпочтительный пример (IV) включает фенилхлорформиат или 1,1'-карбонилдиимидазол. Реакцию проводят в подходящем растворителе, таком как углеводород, в частности, ароматический углеводород, например толуол, или в простом эфире, например ТГФ. Температура реакции зависит от химической активности реагента (IV), но обычно она является комнатной температурой или ниже. В том случае, когда в качестве реагента используется N,N'-карбонилдиимидазол, реакцию предпочтительно проводят при комнатной температуре (т.е. примерно при 25 С). Реакцию обычно завершают после ее протекания в течение времени менее 1 ч, например после ее протекания в течение примерно 1/2 ч. Продукт данной реакции обычно не выделяют; он может быть представлен следующей формулой где Y является таким, как определено выше, и, в частности, представляет собой имидазолильную группу или арилоксигруппу, например фенокси- или замещенную феноксигруппу. Предполагается, что промежуточные продукты формулы (IV-a) являются новыми и составляют дополнительный аспект настоящего изобретения. Предпочтительны промежуточные продукты формулы (IV-a), где Y означает имидазол-1-ил. Продукт предыдущей реакции, т.е. промежуточный продукт формулы (IV-a), обычно не выделяют и сразу же подвергают взаимодействию с амином формулы (V), указанным выше. Предпочтительный амин представляет собой аммиак, но он может быть также аммониевой солью. В таком случае аммиак или аммониевая соль находится в водной среде и добавляется к раствору промежуточного продукта (IV-a) при комнатной температуре. При использовании (V) в водной среде рекомендуется сильное перемешивание из-за наличия двухфазной системы. Реакция завершается через несколько часов, в частности примерно через 4 ч. Отделяют органическую фазу и продукт может быть необязательно выделен и очищен. Органическая фаза с растворенным в ней продуктом (VI) может быть также использована как таковая на последующей реакционной стадии. Стадия с. Получение 2-(замещенный фенил)-2-гидроксиэтилкарбамата (I). Данная реакционная стадия включает удаление защиты для гидроксильной функциональной группы и зависит от природы группы Р. Когда Р является 2-метокси-2-пропилом, удаление осуществляется добавлением соответствующей кислоты, например хлористо-водородной кислоты. При предпочтительном варианте осуществления продукт (VI) используется растворенным в растворителе предыдущей стадии. При перемешивании добавляют воду и концентрированную хлористоводородную кислоту. Уже через несколько минут реакция полностью завершается и конечный продукт(I) начинает осаждаться. Реакционная смесь перемешивается в течение пары часов, в частности в течение около 4 ч, вследствие того, что в таком случае конечный продукт может быть легче отфильтрован. Затем конечный продукт отфильтровывают и промывают. Конечный продукт (I) может быть перекристаллизован из подходящего растворителя, такого как спирт, например метанол, предпочтительно добавлением подкисленной воды. Исходные продукты. Исходные продукты (II) получают способом, который характеризуется:(d) реакцией эстерификации соответствующим образом замещенной миндальной кислоты формулы с получением соответствующего сложного эфира формулы где в (VIII) и (IX) R и R3 являются такими, как определено выше, и R предпочтительно представляет собой 2-хлор и/или R3 предпочтительно представляет собой метил; и(е) обработкой сложного эфира (IX) подходящим реагентом, генерирующим защитную группу для гидрокси, с получением таким образом промежуточного продукта формулы где R и Р являются такими, как определено выше. Сложный эфир (IX) подвергают взаимодействию с соответствующим реагентом, способным вводить защитную группу для гидрокси. Предпочтительный реагент представляет собой 2-метоксипропен. Альтернативно, последовательность вышеуказанных стадий может быть изменена на обратную, т.е. может быть введена защитная группа для гидрокси и затем образован сложный эфир. Стадия (d). Получение сложных эфиров замещенной миндальной кислоты (IX). В соответствии с данной реакционной стадией исходную кислоту (VIII) подвергают взаимодействию в спирте, из которых получают сложный эфир (IX). Обычно используется С 1-4 алканол, предпочтительно метанол, вследствие чего получаются соответствующие сложные С 1-4 алкиловые эфиры или предпочтительно сложный метиловый эфир формулы (VIII). Реакцию проводят с избытком сильной кислоты, предпочтительно галогенводородной кислоты, такой как НСl, в частности с 1-4 мол.экв., предпочтительно с 2,5 мол.экв. концентрированной НСl. Реакция также происходит с каталитическим количеством серной кислоты или также с SOCl2. В последнем случае реакция является сильно экзотермической, что требует принятия соответствующих мер для контроля температуры. Реакцию предпочтительно проводят при комнатной температуре или слегка повышенных температурах, предпочтительно не выше около 30 С. Время реакции обычно составляет менее около 1 ч, например около 30 мин. Образовавшийся сложный эфир (IX) обычно представляет собой маслянистое соединение, которое используется как таковое на последующих стадиях способа. При предпочтительном варианте осуществления способа сложный эфир (IX) сохраняют в растворителе, в котором он получен, и используют затем растворенным в данном растворителе. Стадия (е). Получение сложных эфиров замещенной миндальной кислоты с защищенной гидроксильной группой. Условия реакции данной стадии зависят от природы защитной группы Р. При предпочтительном варианте осуществления Р является 2-метокси-1-пропилом, который получен из 2-метоксипропена. Последний растворяют в подходящем растворителе, в частности в таком растворителе, в котором осуществляют другие реакционные стадии. Полученный раствор добавляют к раствору промежуточного продукта (IX) в подходящем растворителе, который был подкислен добавлением,например, хлористо-водородной кислоты, в частности введением над раствором газообразной хлористоводородной кислоты. рН должен быть предпочтительно низким, например рН 1-2. Растворитель промежуточного продукта (IX) должен быть предпочтительно таким же, как растворитель, в котором растворяли 2-метоксипропен, и более предпочтительно он должен быть таким, как растворитель, использованный на других реакционных стадиях. Реакция завершается в течение времени менее 1 ч, обычно в течение получаса. Способ в соответствии с настоящим изобретением дает конечный продукт (I) с высоким выходом и-7 007741 высокой степенью чистоты и может быть расширен до партий производственного масштаба. Особый аспект настоящего способа состоит в том, что он оставляет нетронутой стереохимическую целостность асимметрического центра в атоме углерода, несущем гидроксильную функциональную группу, т.е. настоящий способ показывает незначительную рацемизацию или ее отсутствие. Различные промежуточные продукты способа, включающего стадии получения исходных продуктов, могут быть выделены и, в случае необходимости, дополнительно очищены перед дальнейшим использованием на следующей стадии. Альтернативно, все стадии способа, в случае необходимости, включающие также стадии получения исходных продуктов, могут быть осуществлены в одном и том же растворителе, т.е. без выделения и необязательной очистки промежуточных продуктов. В последнем случае может быть выгодной отгонка некоторого количества растворителя или добавление некоторого количества растворителя во время одной или нескольких реакционных стадий. Подходящим растворителем для выполнения способа с одним и тем же растворителем является ароматический углеводород, предпочтительно толуол. Можно также осуществлять некоторое число стадий в одном растворителе и другие стадии - в другом растворителе. Проведение всего способа в одном растворителе имеет особое преимущество, состоящее в том, что способ является гораздо более простым и может быть осуществлен более быстро без выбрасывания или регенерации растворителей, что является выгодным с экономической точки зрения, а также с точки зрения охраны окружающей среды. Полиморфные модификации. Настоящее изобретение дополнительно относится к новым кристаллическим структурам соединения формулы (I), в которой R представляет собой 2-хлор, R1 и R2 представляют собой водород, указанное соединение далее будет обозначено как соединение (I-а). Кристаллические формы соединения (I-а) могут быть получены соответствующей перекристаллизацией соединения формулы (I-а) из подходящего органического растворителя. В зависимости от методики перекристаллизации может быть получена или форма А, или форма В. Одна кристаллическая форма соединения (I-а) обозначена как 'форма А' и получена перекристаллизацией соединения (I-а) из подходящего растворителя. В данной методике перекристаллизации поддерживают температуру ниже 60 С, в частности ниже 50 С. Подходящими являются такие растворители, в которых соединение формулы (I-а) растворяется при повышенной температуре и относительно плохо растворимо при пониженной температуре, например при температуре ниже 20 С, или ниже 10 С, или даже ниже 0 С, или -10 С. Подходящие растворители представляют собой низшие алканолы, т.е. С 1-4 алканолы и, в частности, метанол. В одном варианте соединение (I-а) растворяют нагревом или кипячением с обратным холодильником в метаноле или нагревом в низшем алканоле. Температура не должна подниматься выше 60 С. Затем раствор охлаждают, предпочтительно медленно, например, просто позволяя раствору остыть. При температуре около 50 С кристаллизация уже начинается. Смесь перемешивается при комнатной температуре в течение 15-20 ч после добавления подкисленной воды. Воду предпочтительно подкисляют сильной кислотой, например хлористо-водородной кислотой или подобной кислотой, до значения рН от около 3 до 4, в частности до около 3,5. Затем смесь охлаждают примерно до 0-10 С, в частности до около 5 С, и перемешивают при данной температуре в течение длительного периода времени, например в течение около 1 ч. Затем кристаллический конечный продукт фильтруют и сушат. В дополнительном аспекте настоящее изобретение относится к соединению (I-а), находящемуся преимущественно в форме А. Изобретение, в частности, относится к соединению (I-а), находящемуся в виде полиморфной смеси, которая преимущественно содержит форму А. Более конкретно, изобретение относится к соединению (I-а), находящемуся в виде полиморфной смеси, содержащей по меньшей мере 90% или более формы А, в частности 95% или более формы А и наиболее предпочтительно 99% или более формы А. Следовательно, указанная выше перекристаллизация соединения формулы (I-а) дает новую кристаллическую форму соединения (I-а), обозначенную в данном описании 'форма А'. Соединение (I-а) в форме А имеет конкретную кристаллическую форму, т.е. моноклинную кристаллическую форму. Одна кристаллическая форма соединения (I-а) обозначена как 'форма В' и получена перекристаллизацией соединения (I-а) из подходящего растворителя. В данной методике перекристаллизации устанавливают температуру выше 60 С, в частности выше 70 С. Подходящими являются такие растворители, в которых соединение формулы (I-а) растворяется при повышенной температуре и относительно плохо растворимо при пониженной температуре, например при температуре ниже 20 С, или ниже 10 С, или даже ниже 0 С, или -10 С. Подходящие растворители представляют собой низшие алканолы, например,С 3-4 алканолы, и, в частности, пропанол (н-пропанол или 2-пропанол). Другие подходящие растворители представляют собой сложные эфиры, такие как этилацетат или подобный растворитель или их смеси с низкокипящими галогенированными углеводородами, такими как трихлорметан или дихлорметан, который является предпочтительным. В данном случае исходный продукт сначала предпочтительно растворяют в этилацетате, после чего добавляют галогенированный углеводород. Используемый объем галоге-8 007741 нированного углеводорода примерно в пять раз (об/об) превышает объем этилацетата, объемное отношение (этилацетат : галогенированный углеводород) предпочтительно находится в диапазоне от около 1:2 до около 1:5, например, оно может составлять около 1:3. Еще более подходящие растворители представляют собой полиолы, в частности, гликоли, такие как этиленгликоль, который предпочтителен, или пропиленгликоль, бутиленгликоль и подобные. Может быть добавлена вода, в частности, когда в качестве растворителя используются полиолы. Если добавляют воду, она может быть подкислена подходящей сильной кислотой, такой как хлористоводородная кислота, до низкого значения рН, например до рН, который находится в диапазоне рН 2-5, например до рН, который примерно равен 3. Вода может быть добавлена в различных количествах. Так например, когда используются этиленгликоль или подобные гликоли, объемное отношение гликоля к добавленной воде находится в диапазоне от 1:1 до около 1:8 или от 1:2 до 1:5, например, около 1:4 (гликоль: вода, об/об). В одном типе варианта соединение (I-а) растворяют нагревом или кипячением с обратным холодильником в 2-пропаноле, этилацетате или смеси этилацетат/дихлорметан, более предпочтительно в смеси этилацетат/дихлорметан при соотношении указанных компонентов в смеси 1:3. Затем раствору дают возможность охлаждаться, после чего требуемый продукт кристаллизуется. Данная методика может быть использована также для превращения формы А в форму В, т.е. заменой в вышеуказанной методике соединения (I-а) формой А. Форма В может быть получена также в высокочистой форме нагревом формы А до 130 С в течение периода времени 60 мин в виде трехклинных кристаллов (эксперимент с использованием ДСК (DSC. В дополнительном аспекте настоящее изобретение относится к соединению (I-а), находящемуся преимущественно в форме В. Изобретение, в частности, относится к соединению (I-а), находящемуся в виде полиморфной смеси, которая преимущественно содержит форму В. Более конкретно, изобретение относится к соединению (I-а), находящемуся в виде полиморфной смеси, содержащей по меньшей мере 90% или более формы В, в частности 95% или более формы В и наиболее предпочтительно 99% или более формы В. Новые кристаллические формы соединения формулы (I-а) могут быть охарактеризованы их соответствующими рентгенограммами в порошке с использованием соответствующих порошковых дифрактометров, в частности, с применением методики, представленной в примерах. В соответствии с дополнительным аспектом данное изобретение относится к способу получения формы А соединения формулы (I-а) или соединения формулы (I-а) преимущественно в форме А, при этом способ включает растворение соединения формулы (I-а) в подходящем растворителе, нагрев растворителя до температуры ниже около 60 С и выше около 50 С и последующее предоставление возможности данному раствору охлаждаться. Подходящие растворители представляют собой спирты, в частности, С 1-4 алканолы, предпочтительно метанол. В частном варианте осуществления данного способа нагретому раствору дают возможность охлаждаться до появления первых кристаллов, в частности раствору дают возможность охлаждаться примерно до 50 С, обеспечивая таким образом появление первых кристаллов, и затем дают возможность охлаждаться дальше, в частности, до комнатной температуры. В соответствии с другим аспектом данное изобретение относится к способу получения формы В соединения формулы (I-а) или соединения формулы (I-а) преимущественно в форме В, при этом способ включает перекристаллизацию соединения (I-а) из подходящего растворителя при температуре 60 С или выше, в частности при 70 С или выше. Подходящие растворители представляют собой, например, низшие алканолы, например С 3-4 алканолы и, в частности, пропанол. Другие подходящие растворители представляют собой этилацетат или его смеси с низкокипящими галогенированными углеводородами, такими как дихлорметан. Следующие примеры раскрывают данное изобретение более подробно и они предназначены для иллюстрации изобретения, но не для его ограничения. Примеры Получение (S)-(+)-2-(2-хлорфенил)-2-гидроксиэтилкарбамата (соединение (I-а. Пример 1. Сложный метиловый эфир S-(+)-2-хлорминдальной кислоты (промежуточное соединение 1).(S)-(+)-2-Хлорминдальную кислоту (100,0 г, 535,9 ммоль) растворяют при комнатной температуре в метаноле (553,0 г). После охлаждения до 105 С на поверхность раствора в течение 30 мин подают газообразный хлористый водород при поддержании температуры ниже 255 С. За ходом данной реакции и последующих реакций следят с использованием ВЭЖХ. Триметилортоформиат (62,6 г, 589,5 ммоль) при температуре 205 С добавляют к светло-желтому раствору до обесцвечивания, который перемешивают в течение 30 мин при такой же температуре. Затем растворитель и газообразный хлористый водород удаляют в вакууме (405 С, 4010 мбар) насколько это возможно. Маслянистый остаток разбавляют толуолом (130 г) и опять удаляют растворитель в вакууме насколько это возможно. Добавляют толуол (261 г) и раствор охлаждают до 155 С. Пример 2. 2-Метокси-1-пропильное производное сложного метилового эфира (S)-(+)-2-хлорминдальной кислоты (промежуточное соединение 2). На поверхность раствора подают газообразный хлористый водород (0,55 г, 15 ммоль) (значение рН падает от 3 до 1-2). Во второй колбе растворяют в толуоле (120 г) при 205 С 2-метоксипропен (78,0 г,-9 007741 1081,7 ммоль) и добавляют вышеполученный раствор промежуточного соединения 1 при температуре в диапазоне 255 С (приблизительно 30 мин). После завершения добавления смесь перемешивают в течение 30 мин. К светло-желтому раствору добавляют триэтиламин (14,0 г, 138,3 ммоль) до обесцвечивания,его перемешивают при комнатной температуре в течение 5 мин и разбавляют толуолом (402 г) (если не достигается рН 8, добавляют триэтиламин). Смесь промывают водой (1x150 г), водным раствором гидрокарбоната натрия (5%, 1x150 г) и водным насыщенным раствором хлорида натрия (1x150 г). В результате сушки сульфатом натрия (приблизительно 65 г), фильтрации и удаления приблизительно 86 г толуола в вакууме получают промежуточное соединение 2. Пример 3. 2-(2-Хлорфенил)-2-(2-(2-метокси)пропил) этанол (промежуточное соединение 3). Сначала добавляют Venpure ERS В (13,0 г) и затем Venpure ERS A (13,0 г) и смесь нагревают в течение 45 мин до 855 С. Внешний вид раствора меняется от прозрачного до слегка мутного. В раствор добавляют по каплям Venpure ERS С (73,9 г, приблизительно 1239,4 ммоль) таким образом, чтобы установилась температура 955 С (приблизительно 45 мин, возможна задержка начала экзотермической реакции). Реакция заканчивается через 15 мин после завершения добавления по каплям, что показывает также появление в смеси изменения цвета от молочного до серого (за счет цинка). Если вышеуказанное не происходит, в реакционную смесь непосредственно дополнительно добавляют 7,0 г Venpure ERS В и 7,0 г Venpure ERS А, и всю образовавшуюся смесь нагревают до тех пор, пока произойдет завершение конверсии. Раствор охлаждают до 155 С и добавляют метанол (30 г) (происходит выделение водорода). Добавляют по каплям водный раствор гидроксида натрия (30%, 238,0 г) при поддержании температуры ниже 20 С (приблизительно 30 мин, выше 25 С наблюдается интенсивное пенообразование). После завершения добавления двухфазную смесь нагревают в течение 1 ч до 505 С и ранее образованный осадок теперь растворяется. После охлаждения до комнатной температуры фазы разделяют и бесцветную органическую фазу промывают водой (1x200 г) и водным насыщенным раствором хлорида натрия (1x200 г). В результате сушки сульфатом натрия (приблизительно 65 г), фильтрации и удаления в вакууме приблизительно 180 г растворителя получают раствор промежуточного соединения 3. Пример 4. 2-(2-Хлорфенил)-2-(2-(2-метокси)пропил)этилкарбамат (промежуточное соединение 5). К раствору, полученному в примере 3, в течение 30 мин при температуре в диапазоне 255 С добавляют суспензию 1,1'-карбонилдиимидазола (103,8 г, 641,4 ммоль) в толуоле (180 г). Затем завершают конверсию в 1-имидазолил-1'-[2-(2-хлорфенил)-2-(2-(2-метоксипропил]этоксикарбонил (промежуточное соединение 4). Добавляют толуол (200 г) и водный раствор гидроксида аммония (25%, 364 г). После энергичного перемешивания при комнатной температуре в течение 3 ч фазы разделяют и органическую фазу, содержащую промежуточное соединение 5, промывают водой (1x200 г) и водным насыщенным раствором хлорида натрия (1x200 г). Пример 5. Получение соединения (I-а). К органической фазе, содержащей промежуточное соединение 5, полученное как указано выше, добавляют воду (160 г) и концентрированный водный раствор хлористоводородной кислоты (39 г), дающий значение рН 1,0. После перемешивания при комнатной температуре в течение 5 мин начинается осаждение соединения (I-а). Через 4 ч продукт отфильтровывают, фильтровальный осадок промывают водой(3 х 75 г) и толуолом (3 х 44 г). После сушки (505 С, 40+10 мбар) получают 96,9 г соединения (I-а) (степень чистоты 99,8%, определенная анализом ВЭЖХ 99,0%, ее (энант. избыток), определенный хиральной ВЭЖХ, 99,9%, 444,8 ммоль, общий выход 83,0%). Пример 6. Получение соединения (I-а) формы А. Продукт предшествующего примера суспендируют в метаноле (115 г) и после нагрева до температуры перегонки образованный раствор фильтруют горячим, охлаждают до комнатной температуры и перемешивают при указанной температуре в течение 15-20 ч. Добавляют воду (577 мг), содержащую концентрированную хлористо-водородную кислоту (приблизительно 13 мг, значение рН раствора составляет 3,70,2). Смесь охлаждают до 55 С, перемешивают при указанной температуре в течение 2 ч и затем фильтруют. Отфильтрованный осадок промывают водой (3 х 30 г). После сушки (505 С, 5010 мбар) получают 93,0 г соединения (I-а) формы А (степень чистоты 100%, анализ 99,7%, ее 99,9%, 430,0 ммоль, общий выход 80,2%). Пример 7. Определение полиморфной формы рентгеновской порошковой дифрактометрией. Приготовление образца. Примерно от 0,5 г до 1 г образца осторожно прессуют в держателе образца порошка для получения четкой гладкой поверхности. Аппаратура. Используют систему на основе порошкового дифрактометра APD 1700 (Philips), регулируемую компьютером, с автоматическим расширением щели и вторичным монохроматором или эквивалентное оборудование. Условия регистрации. Излучение меди: K1=0,15406 и К 2=0,15444 нм Напряжение: 40 кВ- 10007741 Ток: 30 мА Расширение щели: автоматическое Входная щель: 0,1 Графитовый вторичный монохроматор для Cu-K1Xe пропорционального счетчика Счет начинается при 2 1,5 и осуществляется на шагах от 2 0,02 до достижения 2 40. Время счета в интервале составляет 3 с и общее время регистрации составляет 2 ч 50 мин. Форма А соединения формулы (I-а) может быть охарактеризована ее рентгенограммой в порошке,которая включает основные пики, перечисленные в табл. 1 в примерах. Таблица 1. Пики порошковой дифракции рентгеновских лучей формы А. Форма В соединения формулы (II) может быть охарактеризована ее порошковой рентгенограммой,которая включает основные пики, перечисленные в табл. 2. Форма А. Пример 8. Получение формы В соединения (I-а). 5,0 г (19,0 ммоль) сырого RWJ-333369 (полиморф А) растворяют в 4,75 г 2-пропанола нагревом до температуры перегонки. Образовавшийся раствор охлаждают до комнатной температуры в течение 30- 11007741 мин, после чего вещество кристаллизуется. После дополнительного перемешивания при 4 С в течение 1 ч продукт отфильтровывают и сушат при 80 С и 20 мбар в течение 8 ч. Выход: 4,0 г (15,2 ммоль, 80%),полиморфная форма В. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулыR1 и R2 независимо представляют собой водород или С 1-4 алкил,включающий(а) восстановление сложного эфира формулы где Р представляет собой соответствующую защитную группу для спирта;R3 означает С 1-4 алкил; соответствующим восстановителем сложного эфира до спирта, с получением спирта формулы(b) взаимодействие спирта формулы (III) с карбонильным соединением формулы где X и Y представляют собой соответствующие удаляемые группы; и затем с амином формулы с получением таким образом соединения формулы(с) удаление защитной группы Р с получением соединения формулы I. 2. Способ по п.1, в котором R представляет собой 2-хлор, R1 и R2 представляют собой водород. 3. Способ по п.1 или 2, в котором защитная группа для спирта выбрана из 2-(2-метокси)пропила. 4. Способ по п.1 или 2, в котором карбонильное соединение формулы (IV) выбрано из диимидазолилкарбонила, арил- или замещенный арилгалогенформиата. 5. Способ по п.1 или 2, в котором R3 означает метил. 6. Способ по п.1 или 2, в котором соответствующий восстановитель сложного эфира до спирта представляет собой гидрид металла или комплекс гидрида металла. 7. Соединение формулы где R, R1 и R2 являются такими, как определено в п.1 или 2, и Р является группой типа простого эфира. 8. Соединение по п.7, где группа типа простого эфира выбрана из метоксиметиловых эфиров- 13007741 1-этоксиэтиловых эфиров; 1-метил-1-метоксиэтиловых эфиров; трифениловых эфиров и их соответствующих производных; бензиловых эфиров и их соответствующих производных; 4-метокситетрагидропираниловых эфиров; 4-метокситетрагидротиопираниловых эфиров. 9. Соединение по любому из пп.7 или 8, где Р представляет собой 2-(2-метокси)пропил. 10. Соединение формулы (I-а) отличающееся тем, что соединение формулы (I-а) находится в моноклинной кристаллической форме. 11. Соединение формулы (I-а), как определено в п.10, имеющее следующие пики дифракции рентгеновских лучей в порошке- 14007741 12. Соединение по п.10, по существу, не содержащее другие полиморфные формы соединения формулы (I-а). 13. Соединение по п.11, по существу, не содержащее другие полиморфные формы соединения формулы (I-а). 14. Соединение формулы (I-а) отличающееся тем, что соединение формулы (I-а) находится в триклинной кристаллической форме. 15. Соединение формулы (I-а) по п.14, имеющее следующие пики дифракции рентгеновских лучей в порошке: 16. Соединение по п.14, по существу, не содержащее другие полиморфные формы соединения формулы (I-а). 17. Соединение по п.15, по существу, не содержащее другие полиморфные формы соединения формулы (I-а).
МПК / Метки
МПК: C07C 33/46, C07C 269/04, C07C 69/757, C07C 271/12
Метки: получения, 2-(замещенный, способ, фенил)-2-гидроксиэтилкарбаматов
Код ссылки
<a href="https://eas.patents.su/17-7741-sposob-polucheniya-2-zameshhennyjj-fenil-2-gidroksietilkarbamatov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения 2-(замещенный фенил)-2-гидроксиэтилкарбаматов</a>
2-амино-6-(2, 4, 5-замещенный фенил)пиридины для применения в качестве ингибиторов синтазы оксида азота

Номер патента: 7074
Опубликовано: 30.06.2006
Авторы: Волкманн Роберт Альфред, Лоу Джон Адамс Третий
МПК: A61K 31/4427, A61P 25/00, A61K 31/4418...
Метки: ингибиторов, 5-замещенный, азота, применения, качестве, синтазы, оксида, 2-амино-6-(2, фенил)пиридины
Формула / Реферат:
1. Соединение или его фармацевтически приемлемая соль, которые выбраны из приведенных ниже соединений и их фармацевтически приемлемых солей 6-[4-(N,N-диметиламинометил)-5-этил-2-метоксифенил]пиридин-2-иламин; 6-[4-(N-метиламинометил)-5-этил-2-метоксифенил]пиридин-2-иламин; 6-[4-(3-азетидинокси)-5-этил-2-метоксифенил]пиридин-2-иламин. 2. Соединение по п.1, где указанное соединение представляет собой...
Способ селективного получения z-изомера 3-(2-замещенный-винил) цефалоспорина

Номер патента: 2449
Опубликовано: 25.04.2002
Авторы: Иинума Катсухару, Ивасава Хироюки, Мураи Ясуси, Ватанабе Татсуо, Окада Юмико, Сукегава Масамити
МПК: A61K 31/545, C07D 501/04
Метки: цефалоспорина, z-изомера, селективного, 3-(2-замещенный-винил, способ, получения
Формула / Реферат:
1. Способ селективного получения Z-изомера 7-N-(незамещенной или замещенной амино)-3-[2-(4-замещенный или незамещенный тиазол-5-ил)винил]-3-цефем-4-карбоновой кислоты или ее сложного эфира, представленных следующей общей формулой (IV) где R1 означает атом водорода или одновалентную защитную группу для амино или R1 означает 2-(2-N-защищенный или незащищенный аминотиазол-4-ил)-2-алкоксииминоацетильную группу, имеющую следующую формулу (II) где...
Цефалоспорины, имеющие в положении 7 замещенный бензилоксииминный радикал, способ их получения и применения

Номер патента: 165
Опубликовано: 29.10.1998
Авторы: Асзоди Жозеф, Юмбер Даниель, Фово Патрик
МПК: C07C 239/20, C07D 501/04, A61K 31/545...
Метки: получения, применения, положении, радикал, цефалоспорины, имеющие, способ, бензилоксииминный, замещенный
Формула / Реферат:
1. Цефалоспорины общей формулы (I): изомер син в виде внутрикомплексных солей или солей неорганических или органических кислот или оснований, где R1, R2, R3 и R5, одинаковые или различные, представляют собой атом водорода, атом галогена или радикал, выбранный из группы, состоящей из радикалов: гидроксильный, алкильный, включающий от 1 до 4 атомов углерода, с возможностью замещения одним или несколькими атомами галогена, алкилоксильный,...
Замещенный аналог тетрациклина (варианты) и способ его получения (варианты).

Номер патента: 5118
Опубликовано: 28.10.2004
Авторы: Коуза Дэррелл Дж., Ренни Глен, Нельсон Марк Л.
МПК: A61K 31/65, C07C 237/26, A61P 31/04...
Метки: замещенный, тетрациклина, способ, получения, аналог, варианты
Формула / Реферат:
1. Способ получения 7-, 8-, 9- или 13-замещенного аналога тетрациклина, отличающийся тем, что смешивают реакционноспособный предшественник аналога тетрациклина и реакционноспособный предшественник органического заместителя в присутствии катализатора на основе переходного металла в условиях, при которых образуется аналог тетрациклина, содержащий упомянутый органический заместитель. 2. Способ по п.1, отличающийся тем, что в качестве катализатора...
Способ получения замещенных 4-фенил-4-цианоциклогексановых кислот

Номер патента: 3609
Опубликовано: 26.06.2003
Авторы: Мендельсон Вилфорд, Вебб Кевин, Чен Дзианхао
МПК: C07C 51/08, C07D 303/46
Метки: кислот, получения, способ, 4-фенил-4-цианоциклогексановых, замещенных
Формула / Реферат:
1. Способ получения замещённых 4-фенил-4-цианоциклогексановых кислот формулы I в которой R1 представляет группу -(CR4R5)rR6, в которой алкильные фрагменты могут быть необязательно замещены одним или более галогенами; r = 0-6; R4 и R5 независимо выбраны из водорода или C1-2алкила; R6 представляет C3-6циклоалкил или C4-6циклоалкил, содержащий одну или две ненасыщенные связи, причём циклоалкильные фрагменты могут быть необязательно замещены 1-3...
Предыдущий патент: Процесс получения этиловых эфиров 4 – (8-хлор-5,6-дигидро-11н-бензо-(5,6)-циклогепта-(1,2в) пиридин-11-илиден)-1-пиперидинкарбоновой кислоты (лоратадина)
Следующий патент: Трипептиды, несущие простой гидроксипролиновый эфир замещённого хинолина, предназначенные для ингибирования протеазы ns3 (гепатит с)
Случайный патент: Бытовой прибор с автоматическим отключением