Способ получения небиволола
Номер патента: 22194
Опубликовано: 30.11.2015
Авторы: Фаттори Даниэла, Д'андреа Пьеро, Мауро Сандро, Чиполлоне Амалия












Формула / Реферат
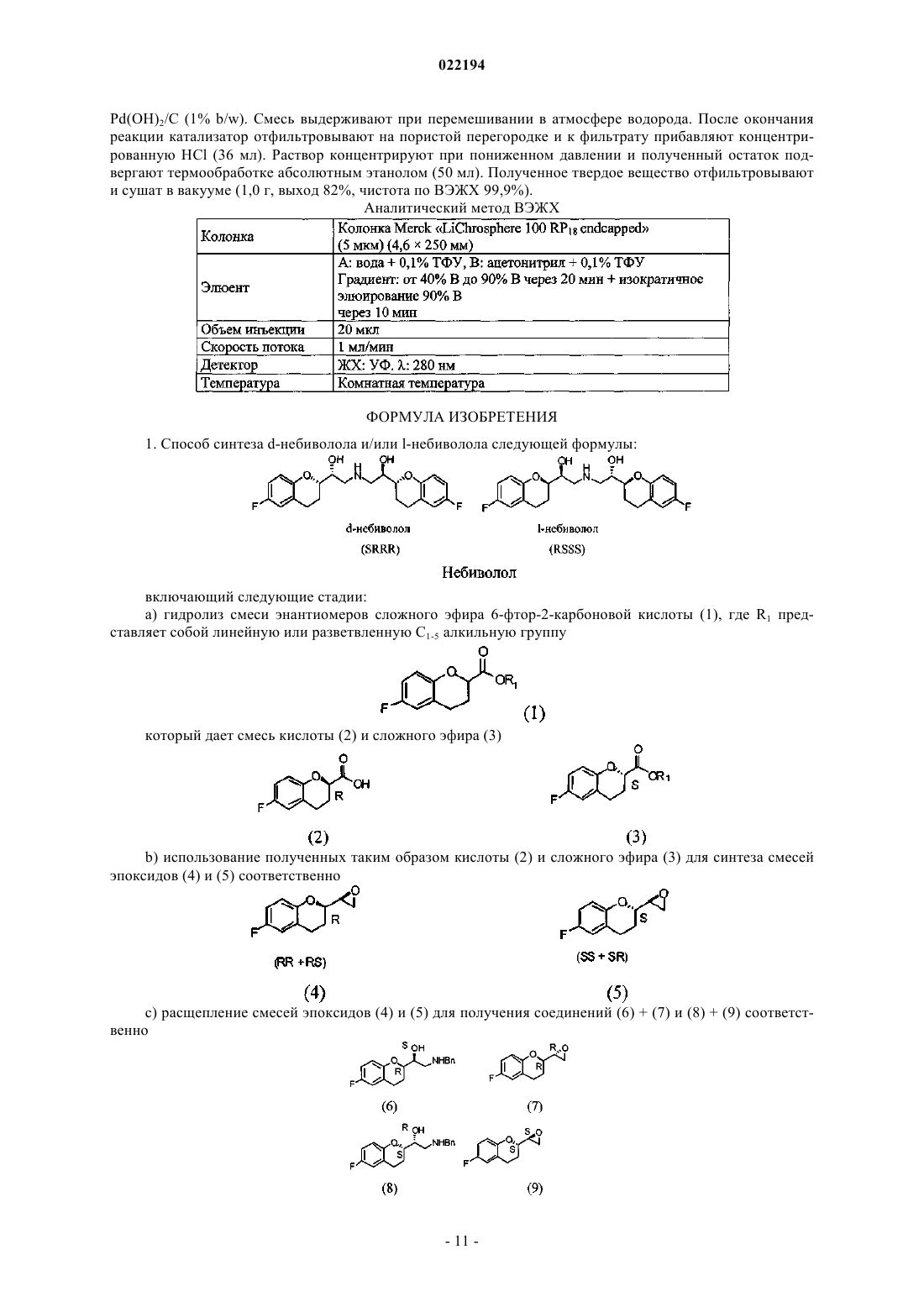
1. Способ синтеза d-небиволола и/или l-небиволола следующей формулы:

включающий следующие стадии:
а) гидролиз смеси энантиомеров сложного эфира 6-фтор-2-карбоновой кислоты (1), где R1 представляет собой линейную или разветвленную С1-5 алкильную группу

который дает смесь кислоты (2) и сложного эфира (3)

b) использование полученных таким образом кислоты (2) и сложного эфира (3) для синтеза смесей эпоксидов (4) и (5) соответственно

с) расщепление смесей эпоксидов (4) и (5) для получения соединений (6) + (7) и (8) + (9) соответственно

d) взаимодействие аминоспиртов (6) и (8) с эпоксидами (7) и (9) для получения l-бензилнебиволола (10) и d-бензилнебиволола (11)

е) удаление бензильной защитной группы,
отличающийся тем, что гидролиз на стадии (а) осуществляют стереоселективной реакцией ферментативного гидролиза, где реакцию ферментативного гидролиза проводят, используя эстеразу, полученную из микроорганизмов рода Ophiostoma, получая смесь R кислоты (2) с энантиомерным избытком >70% и S сложного эфира (3) с энантиомерным избытком >70%; а на стадии (с) кинетическое расщепление смесей эпоксидов (4) и (5) осуществляют путем взаимодействия с бензиламином в пространственно затрудненном спирте.
2. Способ синтеза по п.1, где реакцию ферментативного гидролиза проводят, используя эстеразу, полученную из штамма AJ3 Ophiostoma novo-ulmi.
3. Способ по п.2, где реакцию ферментативного гидролиза проводят при рН от 8 до 11.
4. Способ по п.3, где реакцию ферментативного гидролиза проводят при рН от 8,5 до 10.
5. Способ по п.2, где реакцию ферментативного гидролиза проводят при температуре от 10 до 35°С.
6. Способ по п.5, где реакцию ферментативного гидролиза проводят при температуре от 20 до 25°С.
7. Способ по п.2, где реакцию ферментативного гидролиза проводят в водной среде или в присутствии растворителей, не смешивающихся с водой.
8. Способ по п.2, где ферментативный гидролиз смеси сложных эфиров (1) приводит к образованию смеси (R) кислоты (2) и (S) сложного эфира (3), причем энантиомерный избыток составляет >80% или >90% в обоих компонентах.
9. Способ по п.1, где кислоту (2) превращают в смесь RR+RS эпоксидов (4), тогда как сложный эфир (3) превращают в смесь SS+SR эпоксидов (5).
10. Способ по п.1, где кинетическое расщепление смесей эпоксидов (4) и (5) бензиламином проводят в пространственно затрудненном спирте, выбранном из изопропанола, втор-бутанола, трет-бутанола, 2-метил-2-бутанола, изоамилового спирта, 2-метил-2-пентанола.
11. Способ по п.10, где кинетическое расщепление смеси эпоксидов (4) бензиламином проводят в пространственно затрудненном спирте, выбранном из изопропанола, втор-бутанола, трет-бутанола, 2-метил-2-бутанола, изоамилового спирта, 2-метил-2-пентанола, получая только аминоспирт RS (6), тогда как эпоксид RR (7) выделяют неизмененным.
12. Способ по п.10, где кинетическое расщепление смеси эпоксидов (5) бензиламином проводят в пространственно затрудненном спирте, выбранном из изопропанола, втор-бутанола, трет-бутанола, 2-метил-2-бутанола, изоамилового спирта, 2-метил-2-пентанола, получая только аминоспирт SR (8), тогда как эпоксид SS (9) выделяют неизмененным.
13. Способ по п.11 или 12, где стерически затрудненный спирт представляет собой 2-метил-2-бутанол.
14. Способ по любому из пп.1-13, где аминоспирт RS (6) подвергают взаимодействию с эпоксидом SS (9), получая l-бензилированный небиволол (10); и/или аминоспирт SR (8) подвергают взаимодействию с эпоксидом RR (7), получая d-бензилированный небиволол (11).
15. Способ по п.14, где соединения (10) и (11) смешивают в соотношении 1:1, удаляют защитную бензильную группу и получают конечный продукт небиволол.
16. Способ по пп.1-15, где конечный продукт небиволол подвергают солеобразованию действием хлористо-водородной кислоты, получая соответствующий гидрохлорид.
Текст
Настоящее изобретение относится к новому способу синтеза небиволола, показанному на схеме 1, который содержит уменьшенное число стадий с высоким выходом продукта и характеризуется ферментативным расщеплением предшественника хроманилового сложного эфира. Схема 1(71)(73) Заявитель и патентовладелец: МЕНАРИНИ ИНТЕРНЕШНЛ ОПЕРЕЙШНЗ ЛЮКСЕМБУРГ С.А. Область техники, к которой относится изобретение Настоящее изобретение относится к новому способу синтеза небиволола. Небиволол представляет собой рацемическую смесь двух энантиомеров [2S[2R[R[R ,'-[иминобис(метилен)]бис[6-фторхроман-2-метанол] и [2R[2S[S[S ,'-[имино-бис(метилен)]бис[6-фторхроман-2-метанол]. В частности, сообщают, что ферментативное расщепление исходного хроманилового сложного эфира (1) путем обработки стереоселективным ферментом, принадлежащим к семейству эстераз в нативной или рекомбинантной форме, который может быть получен из Ophiostoma novo-ulmi. Сложные эфиры и кислоты, полученные таким образом, можно превращать в соответствующие"псевдохиральные" эпоксиды, т.е. пары (RR+RS, 4) и (SS+SR, 5), показанные на схеме 1, используя способы, известные специалисту в данной области техники. Схема 1 В свою очередь, компоненты каждой такой пары можно разделять, используя их различную реакционную способность по отношению к бензиламину в растворителе, состоящем из третичного спирта. В этих условиях кинетического расщепления эпоксиды RS и SR превращаются в соответствующие продукты раскрытия цикла (6 и 8), тогда как эпоксиды RR и SS остаются неизмененными. Эпоксид RR (7) затем отделяют от амина RS (6) и эпоксид SS (9) отделяют от амина SR (8) способами, известными специалисту в данной области техники, предпочтительно кристаллизацией основного компонента. Затем амин RS подвергают взаимодействию с эпоксидом SS и получают l-бензилнебиволол. Аналогичным образом амин SR подвергают взаимодействию с эпоксидом RR и получают d-бензилнебиволол.l- и d-Бензилнебивололы, полученные таким образом, объединяют в эквимолярных количествах,кристаллизуют и превращают в небивололHCl (гидрохлорид небиволола) способами, известными специалисту в данной области техники. Предшествующий уровень техники Небиволол известен в качестве антагониста -адренергических рецепторов, антигипертензивного средства, ингибитора агрегации тромбоцитов и вазодилататора. Небиволол обладает основными свойствами и может быть превращен в приемлемую фармацевтическую солевую форму путем обработки кислотой. Гидрохлоридная соль небиволола представляет собой товарную форму. Небиволол содержит 4 асимметрических центра, и поэтому теоретически возможно существование 16 стереоизомеров. Однако из-за своеобразной структуры молекулы (наличие осей симметрии) в действительности могут образоваться только 10 стереоизомеров (схема 2). Схема 2 Возможные изомеры небиволола Действительно, из-за симметрии молекулы возможны RSSS=SSSR, RRSS=SSRR, SRSS=SSRS,RRSR=RSRR, SRSR=RSRS и RRRS=SRRR. Патент США 4654362 (ЕР 0145067, Janssen) описывает синтез продуктов ряда небиволола, в котором используют изомеры эпоксидов в качестве ключевых промежуточных соединений. Продукты иногда получают в виде смеси, иногда в энантиомерно чистом виде без определения абсолютной конфигурации. В частности, в примере 84 указанного патента описано получение смеси изомеров, как определено на схеме 3. Схема 3 Изомеры разделяют на хроматографической колонке на два рацемата эпоксидов (RS/SR, 4/5) и(RR/SS, 7/9). Далее осуществляют раскрытие цикла пары эпоксидов (4 + 5) бензиламином и используют продукты указанной реакции (6 + 8) для раскрытия цикла второй пары эпоксидов (7 + 9). Такая операция приводит к получению 4-бензилированных диастереоизомеров (10-14). В патенте ЕР 0334429 (Janssen) описан тот же способ, что и в патенте ЕР 0145067, но он содержит больше экспериментальных деталей, причем внимание сосредоточено на получении одного изомера небиволола. В этом случае конкретно 6-фторхроманкарбоновую кислоту расщепляют на индивидуальные энантиомеры обработкой (+)-дегидроабиетиламином. Индивидуальные энантиомеры, полученные таким образом, превращают в соответствующие псевдохиральные эпоксиды в соответствии со следующей ниже схемой синтеза (показан изомер S). Схема 4 Описан стереоселективный синтез изомера [2R,S,2'S,'S]-,'-[имино-бис-метилен]бис[6-фтор 3,4-дигидро-2 Н-1-бензопиран-2-метанол]. Используемый способ расщепления сложных эфиров кислот имеет несколько недостатков, касающихся промышленного применения. Действительно, введены дополнительные стадии (образование амидов, фракционированная кристаллизация, гидролиз амидов) и, более того, общий выход довольно низкий. Смесь диастереоизомерных эпоксидов, полученную таким образом, используют в препаративной ВЭЖХ для выделения изомера нужной хиральности. В публикациях международных заявок WO 2006/016376 и WO 2007/083318 компания Hetero DrugsLimited описывает способы фракционированной кристаллизации, применимые для диастереоизомерной смеси (10, 11, 13, 14) бензилнебиволола, которые и в этом случае также приводят к потерям около 50% исходного вещества из-за необходимости удалять нежелательные диастереоизомеры. В публикации международной заявки WO 2004/041805 (Egis Gygyszergyr) описан способ получения рацематов [2S[R[R[R и [2R[S[S[S,'-[имино-бис(метилен)]бис[6-фтор-3,4 дигидро-2 Н-1-бензопиран-2-метанол] и индивидуальных чистых [2S[R[R[R и [2R[S[S[S энантиомеров, исходя из очень непохожих соединений. Число стадий, используемых для синтеза небиволола в виде смеси энантиомеров, составляет около тридцати, что делает такой синтез очень длительным и неэкономичным. Схема 5 В WO 2008/010022 (Cimex Pharma) описан путь, который приводит к синтезу двух энантиомеров небиволола из 6-фторхроманкарбоновых кислот, расщепленных способами, известными из литературы,при этом используют две отдельные последовательности реакций (схема 5 для d-небиволола). При раскрытии эпоксидов бензиламином один продукт раскрытия кристаллизуется из реакционной смеси, но другой диастереоизомер удаляется с маточными растворами, что в этом случае также приводит к удалению значительной доли вещества на уже довольно продвинутой стадии синтеза. Кроме того, последний хиральный центр добавляется на предпоследней стадии восстановлением кетона, довольно чувствительной реакции, в которой для получения оптимальных результатов предусматривается применениеKBH4 и изопропоксида титана. В WO 2008/064826 (Zach System) сообщают о способе расщепления эпоксидов, если пары диастереоизомеров (RS/SR и RR/SS) разделяют хроматографически, путем энантиоселективного раскрытия тех же эпоксидов медиированными хиральными комплексами кобальта II (схема 6). В этом случае необходима стадия хроматографического разделения, не особенно удобная с точки зрения технологичности способа, кроме того, комплексы кобальта требуют осторожности при работе с ними и при утилизации отходов. Схема 6WO 2008/064827 (Zach System) описывает раздельный и энантиоселективный синтез d- и 1 небиволола из двух оптических изомеров защищенного глицеральдегида, такого как 2,2-диметилацеталь(схема 7). Диастереоизомеры разделены известными способами, но они не описаны. Число стадий синтеза больше, чем число стадий классического синтеза, кроме того, альдегидные предшественники известны как не очень стабильные соединения, которые склонны полимеризоваться при хранении в чистом виде и при комнатной температуре. Схема 7 В отношении разделения энантиомеров на уровне 6-фторхроман-2-карбоновой кислоты, известно,что способ образования амида с (+)-дегидроабиетиламином с последующей фракционной кристаллизацией и гидролизом амида для возвращения кислоты (ЕР 0334429) является трудоемким и дает довольно низкие выходы. Что касается ферментативного расщепления сложных эфиров карбоновых кислот, оно представляет собой способ, известный в литературе, но этот способ никогда не использовался для расщепления сложных эфиров фторпроизводных хроман-2-карбоновых кислот и, следовательно, для синтеза небиволола. При этом имеются сообщения об известных примерах, относящихся к хроман-2-карбоксилатам. В патенте США 5037747 сложные эфиры (2R)-гидроксизамещенных бензопиран-2-карбоновых кислот и (2S)-гидроксизамещенных бензопиран-2-карбоновых кислот получают селективным гидролизом соответствующего рацемата, катализируемым липазой из Pseudomonas. Урбан (патент США 5089637 и патент ЕР 0448254) использует ферментативный гидролиз эстеразой, полученной из Pseudomonas fluorescens, для расщепления рацемических смесей общей формулы (I),-4 022194 В WO 96/40975 сообщают о применении микробной эстеразы, полученной из Serratia marcescens для расщепления хроман-2-карбоксилалкилов той же общей формулы, но где RC3. В DE 4430089 описан ряд примеров, в которых сложные эфиры хроман-2-карбоксилаты подвергают ферментативному гидролизу выбранной группой ферментов (химотрипсин, липаза из Candida lipolytica,липаза из Aspergillus oryzae, липаза из Geotrichum candidum, липаза из Aspergillus niger). И наконец, в отношении эстеразы, полученной из аскомицета Ophiostoma novo-ulmi, подробно описаны ее выделение, клонирование в Е. Coli и применение для расщепления сложных эфиров, например,М.Н. Исуповым и соавт. в (М.N. Isupov et al. in Acta Crystallographica - Biological Crystallography SectionD60, p. 1879-1882 (2004 или в ЕР 0687305, тогда как ее применение для расщепления энантиомеров арилалкалоидных кислот и, более конкретно, кетопрофена описано в ЕР 0693134. Литературные данные, имеющиеся к настоящему времени, показывают, что синтез небиволола попрежнему включает в себя многочисленные синтетические проблемы. Первоначальный синтез (Janssen) через эпоксиды (схема 3, смесь 6) несомненно представляет собой более короткий способ, но он требует разделения препаративной ВЭЖХ двух диастереоизомерных пар эпоксидов. Другие способы обычно предусматривают намного больше стадий синтеза. В значительной части описанных способов некоторое количество промежуточных продуктов (которое может достигать 50%), удаляется в отходы для того, чтобы избавиться (исключить) нежелательные диастереоизомеры, неизбежно образующиеся в используемой последовательности синтезов. Таким образом, явно ощущается потребность в разработке нового способа синтеза, подходящего для промышленного применения, в котором не используется хроматографическое разделение, а также существует необходимость в исключении значительных количеств промежуточных соединений за счет сохранения ограниченного числа стадий синтеза. Сущность изобретения В настоящее время неожиданно был открыт более эффективный способ синтеза небиволола (схема 1), который позволяет устранить указанные здесь недостатки известных путей синтеза, т.е. этот способa) устраняет или значительно уменьшает необходимость разделения пар (RR/SS RS/SR) энантиомеров эпоксидов или других диастереомерных промежуточных соединений методом препаративной ВЭЖХ;b) существенно уменьшает потери продукта, представляющего собой нежелательные изомеры и в результате увеличивает общий выход. Обработку смеси двух энантиомеров сложного эфира 6-фторхроман-2-карбоновой кислоты осуществляют грибковой эстеразой (липазой), которую можно получать из грибов вида Ophiostoma. Предпочтительным видом является эстераза из Ophiostoma novo-ulmi, уже описанная в литературе по причине ее стереоселективной активности на сложных эфирах соединений напроксена или кетопрофена. Реакция, проводимая в водной или водной/органической среде, приводит к селективному гидролизу в карбоновую кислоту одного из двух энантиомеров, тогда как другой энантиомер остается в виде сложного эфира. Реакция происходит быстро и с высокой селективностью. Два соединения, полученные таким образом, можно легко разделить кислотно-щелочной экстракцией. Таким образом, целью настоящего изобретения является способ получения небиволола, включающий:a) расщепление (реакцией ферментативного гидролиза) смеси энантиомеров сложного эфира 6 фтор-2-карбоновой кислоты (1), где R1 представляет собой линейную или разветвленную C1-5 алкильную группу, которое дает смесь кислоты (2) и сложного эфира (3) в которой энантиомерный избыток R кислоты (2) составляет 70%, а энантиомерный избыток S сложного эфира (3) составляет 70%; энантиомерный избыток составляет предпочтительно 80% и еще более предпочтительно 90% в обоих компонентахb) использование полученных таким образом кислоты (2) и сложного эфира (3) для синтеза смесей эпоксидов (4) и (5),с.1) реакция кинетического расщепления смесей эпоксидов (4) и (5) бензиламином в пространственно затрудненном спирте для получения соединений (6) + (7) и (8) + (9), соответственно, и их разделение с.2) в качестве альтернативы кинетическому расщеплению, описанному на стадии с.1), хроматографическое разделение смесей эпоксидов (4) и (5) и последующая реакция RS эпоксида с бензиламином дает аминоспирт RS (6), а эпоксид SR с бензиламином дает аминоспирт SR (8).e) снятие защиты с удалением бензильной группы и образованием небиволола Для целей настоящего изобретения группа R1, определенная как линейная или разветвленная С 1-5 алкильная группа, представляет собой радикал, выбранный из метила, этила, пропила, изопропила, бутила, втор-бутила, трет-бутила, амила, трет-амила, предпочтительно радикал, выбранный из метила, этила,пропила, и еще более предпочтительно, если С 1-5 алкильная группа представляет собой этильную группу. Осуществление изобретения Согласно настоящему изобретению небиволол получают способом, описанным на схеме 1, из рацемической смеси сложного эфира 6-фторхроман-2-карбоновой кислоты (1). 6-Фторхроман-2-карбоксилат (1) может быть расщеплен на два его энантиомера с высокой стереоселективностью путем энантиоселективного гидролиза, катализируемого грибковой эстеразой (липазой),которую можно получать из грибов вида Ophiostoma. Предпочтительным видом является эстераза изOphiostoma novo-ulmi, уже описанная в литературе по причине ее стереоселективной активности на сложных эфирах соединений напроксена или кетопрофена. Фермент в виде выделенного кристаллического экспрессированного белка описан М.Н. Исуповым в[М.N. Isupov et al. in Ada Crystallographica - Biological Crystallography Section D60, p. 1879-1882 (2004)]. Фермент описан также в ЕР-В 1-0687305 (WO 94/20634), ЕР 0693134, US 5912164 и ЕР 1626093. Экспрессия фермента в Е coli может быть осуществлена, как описано М.Н. Исуповым и соавт. (ранее) или в ЕР-В 1-0687305 (WO 94/20634). Указанный штамм дает хороший пример активности, однако, принимая во внимание довольно неопределенную природу активности в большом числе родственных штаммов, подразумевается, что объем изобретения не ограничен только этим штаммом. Микроорганизмы вида Ophistoma и их ферментативная активность могут быть использованы для стереоселективного гидролиза рацемического этилового эфира 6-фторхроман-2-карбоновой кислоты, для того, чтобы получить кислоту, значительно обогащенную энантиомером R, например, с 93-100% энантиомерным избытком при конверсии 45-50%, и сохранить в остатке сложный эфир, обогащенный энантиомером S. Таким образом, получается (R) 6-фторхроман-2-карбоновая кислота (2) с энантиомерным избытком 70%, предпочтительно 80% и еще более предпочтительно 90%, при этом (S) 6-фторкарбоновая кислота остается в виде сложного эфира (3) с энантиомерным избытком 70%, предпочтительно 80% и еще более предпочтительно 90%. Реакцию можно проводить на любой смеси энантиомеров, но обычно используют рацемат. Сложный эфир, применяемый для этой реакции, представляет собой эфир 6-фтор-2-карбоновой кислоты (1), где R1 обозначает линейную или разветвленную С 1-5 алкильную группу, выбранную из группы,содержащей метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, амил, трет-амил; предпочтительно из метила, этила, пропила и еще более предпочтительно, если R1 представляет собой этильную группу. Реакцию предпочтительно проводят при рН 8-11, предпочтительно при рН 8,5-10,0. Температура может составлять от 10 до 35 С, но предпочтительно от 20 до 25 С. Реакционная смесь может находиться в водной среде или в присутствии не смешивающихся с водой растворителей. Извлечение обоих соединений возможно способами, известными специалисту в данной области техники, и предпочтительно с помощью ряда кислотно-щелочных экстракций. Оба соединения затем используют для синтеза небиволола (схема 1). Используя способы, известные специалисту в данной области техники (в виде неограничивающего примера, аналогично примеру, описанному в WO 2008/010022), превращают кислоту (2) в диастереоизомерную смесь (RS) и (RR) эпоксидов (4), тогда как сложный эфир (3) превращают в смесь эпоксидов Используя реакцию раскрытия цикла смеси эпоксидов (4) бензиламином в стерически затрудненном спиртовом растворителе (таком как изопропанол, втор-бутанол, трет-бутанол, 2-метил-2-бутанол,изоамиловый спирт, 2-метил-2-пентанол), осуществляют кинетическое расщепление с образованием единственного аминоспирта RS (6), при этом эпоксид RR (7) возвращают неизмененным. Такая же методика, примененная к смеси (5), дает аминоспирт SR (8) и эпоксид SS (9). В качестве альтернативы описанному кинетическому расщеплению смесь эпоксидов (4) может быть хроматографически разделена на два эпоксида RS и RR, а смесь эпоксидов (5) - на два эпоксида SR и SS; затем эпоксид RS подвергают взаимодействию с бензиламином, получая аминоспирт RS (6), тогда как эпоксид SR подвергают взаимодействию с бензиламином, получая аминоспирт SR (8). Наконец, реакция аминоспирта (6) с эпоксидом (7) дает N-бензилированное производное lнебиволола (10), и аналогично реакция аминоспирта (8) с эпоксидом (9) дает N-бензилированное производное d-небиволола (11). Соединения (10) и (11) объединяют в эквимолярных количествах, очищают кристаллизацией (для того, чтобы удалить любые примеси, образованные нежелательными диастереоизомерными соединениями, получаемыми из исходных сложных эфиров/кислот неполной энантиомерной чистоты), дебензилируют и затем образовывают соль, получая целевую конечную соль небиволола. Примеры Изобретение далее подробно описано следующими примерами исключительно для иллюстрации, а не с целью ограничить объем изобретения. Пример 1. Как описано в ЕР-0687305, штамм рекомбинантной Е. Coli, содержащий эстеразу, первоначально экспрессированную в Ophiostoma novo-ulmi, культивируют по методикам, хорошо известным специалисту в данной области техники. Фракцию клеток А подвергают лизису ультразвуком и лизат центрифугируют, получая не содержащий клеток раствор супернатанта. Раствор (1,6 мл), содержащий фермент эстеразу (липазу), полученный из Ophiostoma novo-ulmi (6800 единиц/мл), и суспензию приблизительно 25 г этилового эфира 6-фторхроман-2-карбоновой кислоты (1) в 25 мл деионизированной воды с 100 мкл ТВИН 80, прибавляют к 500 мл 0,1N буферного раствора NaHCO3 (pH 9,7), необязательно доводят уровень рН до 9,7 2N раствором NaOH. Смесь, полученную таким образом, осторожно перемешивают, автоматически поддерживают рН 9,7 путем контролируемого прибавления 2N раствора NaOH. Ход реакции контролируют ВЭЖХ. В конце реакции гидролиза смесь экстрагируют дихлорметаном, чтобы получить сложный эфир в органической фазе. Водный раствор подкисляют 1N соляной кислотой до рН 1 и затем экстрагируют дихлорметаном для извлечения кислоты. Две органические фазы промывают по отдельности насыщенным раствором хлорида натрия и концентрируют, получая соответственно 12,2 г этилового сложного эфира и 11,0 г кислоты. Соотношение энантиомеров (ВЭЖХ):(S) сложный эфир (3)/(R) сложный эфир: 95,31/4,69,(R) кислота (2)/(S) кислота: 95,36/4,64. Оценка удельного вращения в ДМФА при 25 С для смеси кислот (содержащей кислоту (2 показывает, что указанная смесь является левовращающей, что соответствует данным, указанным ЕР 0334429 для R-изомера. Кислота (2) 1 Н-ЯМР (ДМСО-D6, 400 МГц): H (м.д.): 2,04 (2 Н, м, OCHCH2CH2), 2,64 (1 Н, м,OCHCH2CH2), 2,79 (1H, м, OCHCH2CH2), 4,75 (1 Н, т, J = 4,5 Гц, OCHCO), 6,80-7,00 (3 Н, м, CHar), 13,00 Пример 2. Получение производного кислоты Мелдрума Выделенную resolved (R) кислоту (28 г) растворяют в 250 мл безводного дихлорметана; к полученному раствору прибавляют по каплям 1,4 экв. оксалилхлорида и ДМФА. Раствор выдерживают при перемешивании при комнатной температуре и в атмосфере N2; через 1,5 ч растворитель упаривают, получая масло, которое повторно растворяют в 200 мл безводного дихлорметана. Отдельно кислоту Мельдрума (1,05 экв.) и пиридин (2 экв.) растворяют в безводном дихлорметане (150 мл) и оставляют при перемешивании при 0 С в течение 15 мин. К этому раствору прибавляют ранее образованный хлорид кислоты. В конце прибавления смесь оставляют при перемешивании при 0 С в течение 1 ч и при комнатной температуре в течение 45 мин. Затем разбавляют 500 мл дихлорметана и органическую фазу промывают Н 2 О (2200 мл), 2N раствором HCl (100 мл), водой и насыщенным раствором хлорида натрия и сушат над Na2SO4. Получают масло, которое обрабатывают 20 объемами диизопропилового эфира, получая коричневое твердое вещество (40 г, чистота 81% по ВЭЖХ, = 280 нм), которое отфильтровывают и сушат. Полученное твердое вещество используют в последующей реакции без дополнительной очистки. Пример 3. Получение -кетоэфира Неочищенное ацильное производное Мелдрума (R) (40 г) помещают при перемешивании в 110 мл трет-бутанола; полученную смесь нагревают до 80 С в течение 1 ч до тех пор, пока ВЭЖХ показывает исчезновение исходного продукта. После окончания реакции, трет-бутанол выпаривают при пониженном давлении; вносят 500 мл этилацетата и органическую фазу промывают насыщенным раствором NaHCO3,H2O до нейтральной среды, насыщенным раствором хлорида натрия и сушат над Na2SO4. Затем растворитель испаряют, получая 28 г неочищенного -кетоэфира (чистота 69% по ВЭЖХ, = 280 нм) в виде масла, которое используют в последующей реакции без дополнительной очистки. Пример 4. Получение хлоркетоэфира Неочищенный -кетоэфир (R) (28 г) растворяют в 250 мл этилацетата и к полученному раствору прибавляют 0,26 экв. Mg(ClO4)2. Через 30 мин прибавляют 0,95 экв. N-хлорсукцинимида 2 ч. После прибавления полученную смесь перемешивают в течение 1 ч при комнатной температуре. Затем образовавшееся твердое вещество удаляют, прозрачный раствор переносят в делительную воронку, после разбавления 350 мл этилацетата органическую фазу промывают насыщенным раствором хлорида натрия, Н 2 О и сушат над Na2SO4. Растворитель испаряют, получая 34 г неочищенного хлорпроизводного (чистота 79,40% по ВЭЖХ,= 280 нм), который используют в последующей реакции без дополнительной очистки. Пример 5. Получение -хлоркетона Неочищенный хлоркетоэфир (R) (34 г) кипятят с обратным холодильником с НСООН (100 мл),СН 3 СООН (120 мл) и Н 2 О (30 мл); через 1,5 ч контроль ВЭЖХ указывает на окончание реакции. Смесь упаривают при пониженном давлении, прибавляют этилацетат и органическую фазу промывают насыщенным раствором хлорида натрия, насыщенным раствором NaHCO3, H2O и сушат над Na2SO4. Затем растворитель испаряют при пониженном давлении, получая 21 г -хлоркетона (чистота = 60% по ВЭЖХ, = 280 нм) в виде масла, который используют "таким как есть" на следующей стадии без дополнительной очистки. Масло, полученное предшествующей реакцией (21 г), растворяют в 15 об. МеОН, к раствору прибавляют шпателем 2,0 экв. NaBH(ОСОСН 3)3 и перемешивают магнитной мешалкой при комнатной температуре. Через 45 мин прибавляют еще 1 экв. NaBH(ОСОСН 3)3. Через 1 ч после последнего прибавления контроль ВЭЖХ указывает на окончание реакции. Растворитель испаряют при пониженном давлении, вс переносят в делительную воронку с этилацетатом, органическую фазу промывают Н 2 О и насыщенным раствором хлорида натрия и сушат над Na2SO4. Получают 21 г масла, которое очищают флэшхроматографией (отношение силикагель/неочищенное масло: 30:1, элюент: петролейный эфир/AcOEt 92: 8), получая 14,2 г -хлорспирта (чистота = 86,5% по ВЭЖХ,= 280 нм). Пример 7. Получение (RR+RS) эпоксидов (4)-Хлорспирт (14 г) растворяют в 10 об. безводного Et2O и к этому раствору прибавляют 2,8 г NaH,предварительно промытого петролейным эфиром. Через 1 ч контроль ТСХ (силикагель, элюент: петролейный эфир/AcOEt 85:15) указывает на исчезновение исходного хлорспирта (одно пятно на ТСХ) и образование двух эпоксидов (два четко различающихся пятна на ТСХ). Затем реакционную смесь разбавляют 30 об. Et2O и все выливают в 100 мл 1 М раствора NaHSO4, поддерживая сильное перемешивание. Органическую фазу промывают NaHCO3, Н 2 О, насыщенным раствором хлорида натрия и сушат надNa2SO4. Затем растворитель испаряют при пониженном давлении, получая 11,4 г смеси эпоксидов в виде масла (чистота 98% по ВЭЖХ,= 280 нм) в соотношении 51:48. Наличие только двух основных пиков в соотношениях, указанных в анализе с помощью хиральной ВЭЖХ, показывает отсутствие рацемизации в последовательности реакций от (R) кислоты к смеси диастереоизомерных (RR + RS) эпоксидов, с выраженным сохранением стереоцентральной хиральности. Смесь (SR + SS) эпоксидов (5) получают по методике, аналогичной описанной в примерах 2-7, исходя из сложного эфира (3) после его гидролиза в соответствующую кислоту. В этом случае оценка удельного вращения в ДМФА при 25 С для кислоты, таким образом полученной, показывает, что она правовращающая, что соответствует данным, описанным в заявке ЕР 0334429 А 1 для изомера S. Пример 8. Кинетическое расщепление смеси эпоксидов (SS + SR). Раствор смеси (SS +SR) эпоксидов (4,50 г, 22,5 ммоль) и бензиламин (3,8 мл, 35 ммоль) в 2-метил-2 бутаноле (38 мл) перемешивают при комнатной температуре 12 ч. После окончания реакции образовавшийся (SR) амин (8) отфильтровывают в вакууме и сушат (1,90 г, 6,30 ммоль). Фильтрованный раствор выливают в циклогексан (250 мл) и раствор, полученный таким образом, промывают 1M растворомNaHSO4 (100 мл) и Н 2 О (50 мл 2), затем концентрируют при пониженном давлении, получая 1,30 г(6,00 ммоль) (SS) эпоксида (9). Кинетическое расщепление смеси (RS + RR) эпоксидов проводят аналогично расщеплению, описанному в примере 8. Пример 9. Синтез l-бензилнебиволола (SSSR).(6 мл) и смесь кипятят с обратным холодильником до исчезновения исходных реагентов. После окончания реакции смесь оставляют до достижения комнатной температуры и удаляют растворитель при пониженном давлении. Пример 10. Синтез d-бензилнебиволола (RRRS). Соединение (SR)-2-бензиламино-l-(6-фторхроман-2-ил)этанол и (RR) эпоксид обрабатывают, как описано в примере 9, и получают d-бензилнебиволол. Пример 11. Синтез d,l-бензилнебиволола.(3,00 г), объединяют и смесь, полученную таким образом (6,0 г), очищают кристаллизацией, получая 5,0 г N-бензилнебиволола (83%, чистота по ВЭЖХ = 99,6%). Во время очистки кристаллизацией устраняются также примеси, состоящие из нежелательных изомеров, получаемых при неполном энантиоселективном гидролизе исходного этилового эфира 6-фторхроман-2-карбоновой кислоты (1). Пример 12. Синтез гидрохлорида небиволола. Соединение d,l-бензилнебиволол (5,0 г, 410 ммоль) растворяют в метаноле (400 мл) вместе с 20%Pd(OH)2/C (1% b/w). Смесь выдерживают при перемешивании в атмосфере водорода. После окончания реакции катализатор отфильтровывают на пористой перегородке и к фильтрату прибавляют концентрированную HCl (36 мл). Раствор концентрируют при пониженном давлении и полученный остаток подвергают термообработке абсолютным этанолом (50 мл). Полученное твердое вещество отфильтровывают и сушат в вакууме (1,0 г, выход 82%, чистота по ВЭЖХ 99,9%). Аналитический метод ВЭЖХ включающий следующие стадии: а) гидролиз смеси энантиомеров сложного эфира 6-фтор-2-карбоновой кислоты (1), где R1 представляет собой линейную или разветвленную С 1-5 алкильную группу который дает смесь кислоты (2) и сложного эфира (3)b) использование полученных таким образом кислоты (2) и сложного эфира (3) для синтеза смесей эпоксидов (4) и (5) соответственно с) расщепление смесей эпоксидов (4) и (5) для получения соединений (6) + (7) и (8) + (9) соответственно е) удаление бензильной защитной группы,отличающийся тем, что гидролиз на стадии (а) осуществляют стереоселективной реакцией ферментативного гидролиза, где реакцию ферментативного гидролиза проводят, используя эстеразу, полученную из микроорганизмов рода Ophiostoma, получая смесь R кислоты (2) с энантиомерным избытком 70% и S сложного эфира (3) с энантиомерным избытком 70%; а на стадии (с) кинетическое расщепление смесей эпоксидов (4) и (5) осуществляют путем взаимодействия с бензиламином в пространственно затрудненном спирте. 2. Способ синтеза по п.1, где реакцию ферментативного гидролиза проводят, используя эстеразу,полученную из штамма AJ3 Ophiostoma novo-ulmi. 3. Способ по п.2, где реакцию ферментативного гидролиза проводят при рН от 8 до 11. 4. Способ по п.3, где реакцию ферментативного гидролиза проводят при рН от 8,5 до 10. 5. Способ по п.2, где реакцию ферментативного гидролиза проводят при температуре от 10 до 35 С. 6. Способ по п.5, где реакцию ферментативного гидролиза проводят при температуре от 20 до 25 С. 7. Способ по п.2, где реакцию ферментативного гидролиза проводят в водной среде или в присутствии растворителей, не смешивающихся с водой. 8. Способ по п.2, где ферментативный гидролиз смеси сложных эфиров (1) приводит к образованию смеси (R) кислоты (2) и (S) сложного эфира (3), причем энантиомерный избыток составляет 80% или 90% в обоих компонентах. 9. Способ по п.1, где кислоту (2) превращают в смесь RR+RS эпоксидов (4), тогда как сложный эфир (3) превращают в смесь SS+SR эпоксидов (5). 10. Способ по п.1, где кинетическое расщепление смесей эпоксидов (4) и (5) бензиламином проводят в пространственно затрудненном спирте, выбранном из изопропанола, втор-бутанола, трет-бутанола,2-метил-2-бутанола, изоамилового спирта, 2-метил-2-пентанола. 11. Способ по п.10, где кинетическое расщепление смеси эпоксидов (4) бензиламином проводят в пространственно затрудненном спирте, выбранном из изопропанола, втор-бутанола, трет-бутанола, 2 метил-2-бутанола, изоамилового спирта, 2-метил-2-пентанола, получая только аминоспирт RS (6), тогда как эпоксид RR (7) выделяют неизмененным. 12. Способ по п.10, где кинетическое расщепление смеси эпоксидов (5) бензиламином проводят в пространственно затрудненном спирте, выбранном из изопропанола, втор-бутанола, трет-бутанола, 2 метил-2-бутанола, изоамилового спирта, 2-метил-2-пентанола, получая только аминоспирт SR (8), тогда как эпоксид SS (9) выделяют неизмененным. 13. Способ по п.11 или 12, где стерически затрудненный спирт представляет собой 2-метил-2 бутанол. 14. Способ по любому из пп.1-13, где аминоспирт RS (6) подвергают взаимодействию с эпоксидомSS (9), получая l-бензилированный небиволол (10); и/или аминоспирт SR (8) подвергают взаимодействию с эпоксидом RR (7), получая d-бензилированный небиволол (11). 15. Способ по п.14, где соединения (10) и (11) смешивают в соотношении 1:1, удаляют защитную бензильную группу и получают конечный продукт небиволол. 16. Способ по пп.1-15, где конечный продукт небиволол подвергают солеобразованию действием хлористо-водородной кислоты, получая соответствующий гидрохлорид.
МПК / Метки
МПК: C07D 311/58
Метки: получения, небиволола, способ
Код ссылки
<a href="https://eas.patents.su/13-22194-sposob-polucheniya-nebivolola.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения небиволола</a>
Способ получения небиволола

Номер патента: 19691
Опубликовано: 30.05.2014
Авторы: Чиполлоне Амалия, Бартоли Сандра, Фаттори Даниэла
МПК: C07D 311/58
Метки: небиволола, способ, получения
Формула / Реферат:
1. Способ получения Небиволола в форме рацемической смеси двух энантиомеров [2S[2R[R[R]]]] α,α'-[имино-бис-(метилен)] бис-[6-фтор-хроман-2-метанола] и [2R[2S[S[S]]]] α,α'-[имино-бис-(метилен)] бис-[6-фтор-хроман-2-метанола], имеющих следующие формулы:содержащий следующие стадии, на которых:а) смесь четырех изомеров SR, RS, RR и SS эпоксида формулы 1вводят в реакцию с амином R-NH2, где R представляет собой защитную группу,...
Способ получения дихлорпропанола, способ получения эпихлоргидрина, способ получения эпоксидных смол и применение оборудования, обладающего коррозионной стойкостью, в способе получения дихлорпропанола

Номер патента: 14241
Опубликовано: 29.10.2010
Авторы: Франк Кристиан, Краффт Филипп, Бальтазар Доминик, Жильбо Патрик, Сметс Валентин
МПК: C07C 29/62, B01J 19/02, C07C 31/36...
Метки: стойкостью, способе, применение, смол, эпоксидных, обладающего, способ, коррозионной, получения, эпихлоргидрина, оборудования, дихлорпропанола
Формула / Реферат:
1.Способ получения дихлорпропанола, содержащий:(a) стадию, на которой глицерин или сложный эфир глицерина или их смесь вводят во взаимодействие с агентом хлорирования, содержащим хлороводород,(b) по меньшей мере одну другую стадию, осуществляемую на оборудовании, выполненном или имеющем покрытие из материалов, обладающих стойкостью по отношению к агенту хлорирования, в условиях осуществления этой стадии,причем другая стадия является стадией...
Катализатор для получения сложных эфиров,способ получения сложного эфира и способ получения сложного полиэфира с участием такого катализатора

Номер патента: 11171
Опубликовано: 27.02.2009
Авторы: Хэнратти Алан Джозеф, Макинтош Кэлам Гарри, Партридж Мартин Грэхэм
МПК: C08G 63/85, B01J 31/04
Метки: получения, сложных, сложного, эфира, катализатора, эфиров,способ, полиэфира, такого, катализатор, участием, способ
Формула / Реферат:
1. Катализатор для получения сложного эфира в реакции этерификации, состоящий из продукта взаимодействия: a) соединения титана, циркония или гафния; b) 2-оксикарбоновой кислоты; c) четвертичного аммониевого соединения, выбранного из группы, состоящей из гидроксида тетраэтиламмония и гидроксида тетраметиламмония, и d) соединения цинка. 2. Катализатор по п. 1, в котором соединением титана, циркония или гафния является алкоголят, имеющий формулу...
Способ получения дихлорпропанола, способ получения эпихлоргидрина и способ получения эпоксидных смол

Номер патента: 13681
Опубликовано: 30.06.2010
Авторы: Краффт Филипп, Жильбо Патрик
МПК: C07C 31/42, C07C 29/62, C07C 31/36...
Метки: дихлорпропанола, эпихлоргидрина, эпоксидных, получения, смол, способ
Формула / Реферат:
1. Способ получения дихлорпропанола, в котором вводят во взаимодействие глицерин, или сложный эфир глицерина, или их смесь, общее содержание металлов в которых, выраженное в расчете на элементы, выше или равно 0,1 мкг/кг и ниже или равно 1000 мг/кг, и агент хлорирования.2. Способ по п.1, в котором общее содержание металлов ниже или равно 500 мг/кг и который характеризуется по меньшей мере одним из следующих признаков:содержание железа в...
Способ получения композиции сложного полиэфира, полученная композиция, содержащая ее пленка, раствор для получения композиции и способ его получения

Номер патента: 14016
Опубликовано: 30.08.2010
Авторы: Хельдманн Карл-Хайнц, Зайдель Экхард, Отто Бригитта
МПК: C08K 5/00, C08K 5/098, C08J 5/18...
Метки: композиции, способ, получения, сложного, композиция, пленка, полиэфира, раствор, полученная, содержащая
Формула / Реферат:
1. Способ получения композиции сложного полиэфира, включающий следующие стадии:A) этерификацию дикарбоновой кислоты алкандиолом или переэтерификацию диалкилового эфира дикарбоновой кислоты алкандиолом;B) предварительную поликонденсацию полученного диалкилового эфира дикарбоновой кислоты до форполиконденсата;C) поликонденсацию в расплаве форполиконденсата до сложного полиэфира, причем смесь, которую подвергают поликонденсации, содержит по меньшей...
Предыдущий патент: Сухая грануляция металлургического шлака
Следующий патент: Способ удаления пахучих веществ из белья в стирально-сушильной машине и стирально-сушильная машина для осуществления способа
Случайный патент: Способ получения пористого материала на основе углерода