Способ получения циклопропилацетилена.
Номер патента: 74
Опубликовано: 25.06.1998
Авторы: Корли Эдвард Г., Томпсон Эндрю С., Хантингтон Марта











Формула / Реферат
1. Способ получения циклопропилацетилена, включающий следующие стадии:
(а) смешивание, по крайней мере, около 1,0 эквивалента сильного основания, выбранного из группы, включающей н-бутиллитий, амид натрия, диэтиламид натрия, гидрид натрия, гидрид калия, бис(триметилсилил) амид натрия, бис(триметилсилил)амид калия, диизопропиламид лития, втор-бутиллитий, трет-бутиллитий и тетраметилпиперидид лития, в апротонном растворителе с одним эквивалентом 5-галоген-1-пентина в апротонном растворителе при температуре в интервале между около -20 и около 150°С;
(б) повышение температуры реакционной смеси до температуры от около 0 до около 150°С и поддержание температуры в пределах данного интервала, по крайней мере, в течение 15 мин или до тех пор, пока циклизация по существу не завершится; и
(в) гашение реакции любым источником протонов.
2. Способ получения циклопропилацетилена, включающий следующие стадии:
(а) смешивание, по крайней мере, около 1,0 эквивалента сильного основания, выбранного из группы, включающей н-бутиллитий, амид натрия, диэтиламид натрия, гидрид натрия, гидрид калия, бис(триметилсилил) амид натрия, бис(триметилсилил)амид калия, диизопропиламид лития, втор-бутиллитий, трет-бутиллитий и тетраметилпиперидид лития, в апротонном растворителе с одним эквивалентом 5-галоген-1-пентина в апротонном растворителе при температуре в интервале между около -20 и около 150°С;
(б) повышение температуры реакционной смеси до температуры от около 0 до около 150°С и поддержание температуры в пределах данного интервала, по крайней мере, в течение 15 мин или до тех пор, пока циклизация по существу не завершится;
(в) охлаждение реакционной смеси до температуры в интервале между около -30 и около 50°С;
(г) гашение реакции любым источником протонов.
3. Способ по п.2, дополнительно включающий очистку циклопропилацетилена.
4. Способ по п.1, где сильным основанием является н-бутиллитий.
5. Способ по любому из пп.1-3, где апротонный растворитель выбран из тетрагидрофурана, 2,5-диметилтетрагидрофурана, 1,4-диоксана, трет-бутилметилового эфира, диэтоксиметана, диметоксиэтана, циклогексана, гексана и смеси гексана с тетраметилендиамином.
6. Способ по п.5, где апротонным растворителем является циклогексан.
7. Способ по любому из пп.1-3, где источник протонов выбран из насыщенного NH4Cl, НСl и Н2SO4.
8. Способ по любому из пп.1-3, где 5-галоген-1-пентином является 5-хлор-1-пентин.
9. Способ получения циклопропилацетилена, включающий следующие стадии:
(а) смешивание от около 2,0 до около 2,5 эквивалентов н-бутиллития в циклогексане с одним эквивалентом 5-хлор-1-пентина в циклогексане при около 0°С;
(б) нагревание смеси до температуры около 75°С и поддержание данной температуры в течение 5 ч или до тех пор, пока циклизация по существу не завершится;
(в) охлаждение реакционной смеси до температуры около 0°С;
(г) гашение реакции насыщенным NH4Cl.
10. Способ по п.9, дополнительно включающий очистку циклопропилацетилена.
Текст
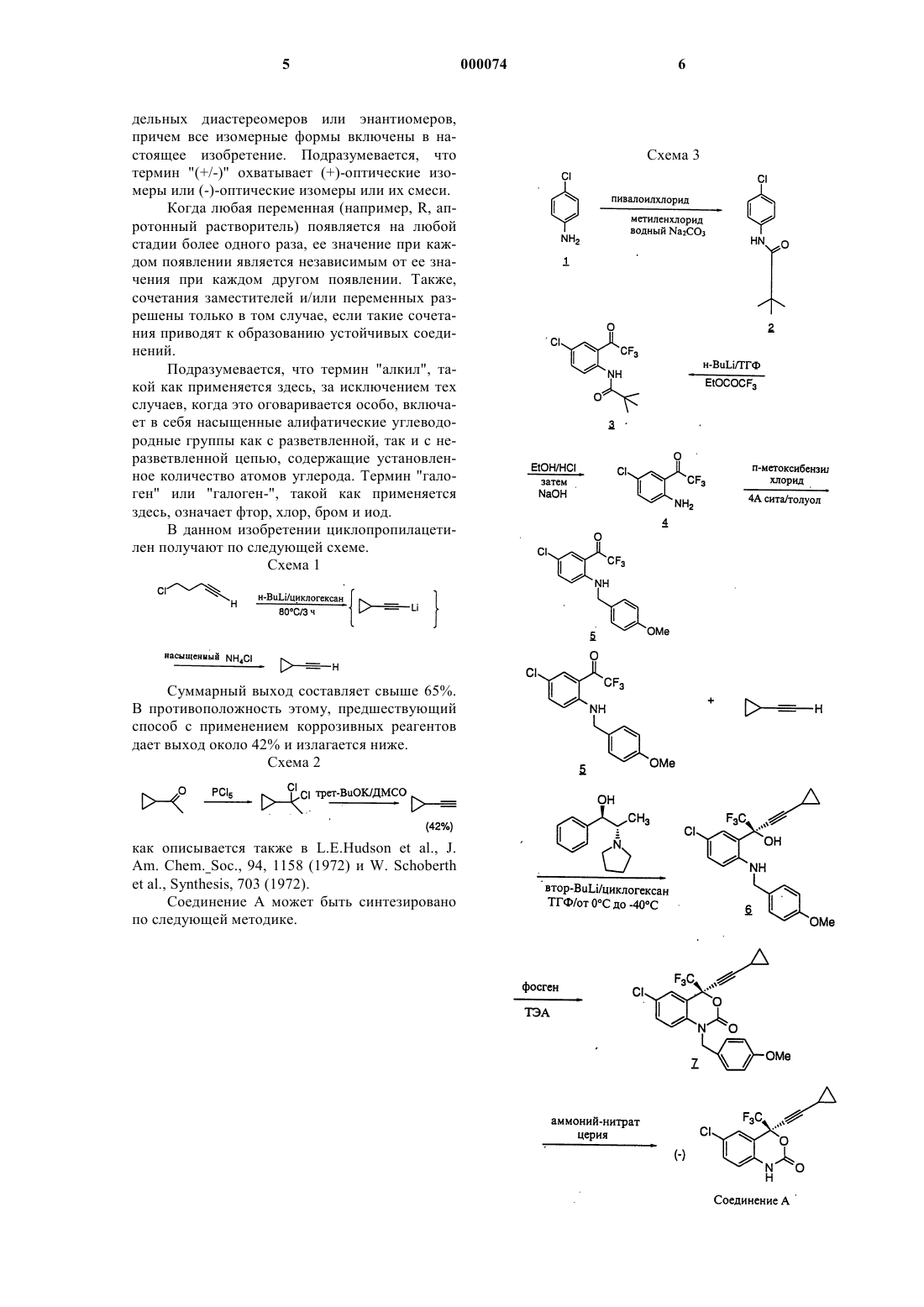
1 Данная заявка 187931 В представляет собой частичное продолжение 187931 А, которая, в свою очередь, представляет собой частичное продолжение заявки Merck 18793, поданной 7 августа 1992 г., U.S.S.N. 07/926607, и заявки 19344. Ретровирус, обозначаемый как вирус иммунодефицита человека (ВИЧ), является этиологическим агентом комплексного заболевания,которое включает в себя прогрессирующую деструкцию иммунной системы (синдром приобретенного иммунодефицита; СПИД) и дегенерацию центральной и периферической нервной системы. Ранее данный вирус был известен какLAV, HTLV-III, или ARV. Характерным признаком репликации ретровируса является обратная транскрипция генома РНК посредством кодируемой вирусом обратной транскриптазы с получением ДНК-копий последовательностей ВИЧ, необходимый этап в репликации вируса. Известно, что некоторые соединения являются ингибиторами обратной транскриптазы и эффективными средствами для лечения СПИД и подобных заболеваний, например азидотимидин или АЗТ. Установление нуклеотидной последовательности ВИЧ показывает наличие гена ро 1 в одной открытой рамке считывания [Ratner, L. etal., Nature, 313, 277 (1985)]. Гомология аминокислотных последовательностей делает очевидным, что последовательность ро 1 кодирует обратную транскриптазу, эндонуклеазу и протеазу ВИЧ [Toh, Н. et al., EMBO J., 4, 1267 (1985);Pearl, L.H., et al., Nature, 329, 351 (1987)]. Заявители предполагают существенно усовершенствованный синтез ингибитора обратной транскриптазы ВИЧ структуры называемого как (-)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2 Н-3,1 бензоксазин-2-он, далее по тексту "Соединение А". Данное соединение чрезвычайно действенно, даже против обратной транскриптазы ВИЧ,устойчивой к другим противовирусным антиВИЧ соединениям. Заявители предложили существенно усовершенствованный синтез циклопропилацетилена, промежуточного продукта для получения Соединения А. Предшествующие способы включали в себя двухстадийные процедуры с применением коррозивных реагентов и давали низкий суммарный выход. Смотри, например,Militzer, Н.С. et al., Synthesis, 998 (1993); Schoberth, W. et al., Synthesis, 703 (1972) [РСl5 и основание]; Sherrod, et al., J. Am. Chem. Soc., 93:8,1925-1940 (апрель 1971 года) [12 в гидразоне; 2 том XXII, 195-201 (1952) [дибромид]. Напротив,настоящий способ является более коротким, чем предшествующие способы, не требует применения коррозивных реагентов и позволяет получать такой же или более высокий, чем в предшествующих способах, выход. Настоящий способ включает в себя циклизацию 5-галоген-1 пентина в сильном основании. Заявители обнаружили, что для успешного исхода реакции требуется образование переходного дианиона, который циклизуется в циклопропилацетилен. Ни один из способов из данной области не позволяет получить дианион. Иначе говоря, работы в данной области указывают на то, что идет множество побочных реакций,включая замещение хлора основанием или депротонированным ацетиленом или галогенметаллический обмен хлорида. Описывается усовершенствованный синтез циклопропилацетилена, промежуточного продукта для получения cоединения А. Синтез включает в себя циклизацию 5-галоген-1 пентина в сильном основании. Соединение А может применяться для ингибирования обратной транскриптазы ВИЧ (и ее устойчивых разновидностей) , предотвращения ВИЧ-инфекции,лечения ВИЧ-инфекции или лечения СПИД и/или САК, так же как и соединения, фармацевтически приемлемые соли (когда это подходит),ингредиенты фармацевтических композиций,так или иначе в сочетании с другими противовирусными средствами, антибактериальными средствами, иммуномодуляторами, антибиотиками или вакцинами. Также описываются способы лечения СПИД, методы предотвращения ВИЧ-инфекции и методы лечения ВИЧинфекции. Способ по настоящему изобретению касается получения циклопропилацетилена, реагента, используемого для введения циклопропилацетиленовой группы во множество антивирусных средств и других соединений медицинского интереса, особенно ингибиторов обратной транскриптазы ВИЧ. В данном изобретении способ получения циклопропилацетилена включает в себя следующие стадии:(а) смешивание, по крайней мере, около 1,0 эквивалента сильного основания в апротонном растворителе с одним эквивалентом 5 галоген-1-пентина в апротонном растворителе при температуре в интервале между приблизительно -20 и приблизительно 150 С;(б) повышение температуры реакционной смеси до интервала между приблизительно 0 и приблизительно 150 С и поддержание температуры в пределах интервала, по крайней мере, в течение 15 мин или до тех пор, пока циклизация, по существу, не завершится;(в) гашение реакции любым источником протонов. 3 В одном осуществлении настоящего изобретения способ получения циклопропилацетилена включает в себя следующие стадии:(а) смешивание, по крайней мере, около 1,0 эквивалента сильного основания в апротонном растворителе с одним эквивалентом 5 галоген-1-пентина в апротонном растворителе при температуре в интервале между приблизительно -20 и приблизительно 150 С;(б) повышение температуры реакционной смеси до интервала между приблизительно 0 и приблизительно 150 С и поддержание температуры в пределах интервала, по крайней мере, в течение 15 мин или до тех пор, пока циклизация, по существу, не завершится;(в) охлаждение реакционной смеси до температуры в интервале между приблизительно(г) гашение реакции любым источником протонов. В другом осуществлении настоящего изобретения добавляется дополнительная стадия очистки желаемого продукта циклопропилацетилена. Одним осуществлением настоящего изобретения является способ получения циклопропилацетилена, который включает в себя следующие стадии:(а) смешивание от приблизительно 2,0 до приблизительно 2,5 эквивалентов н-бутиллития в циклогексане с одним эквивалентом 5-хлор-1 пентина в циклогексане при приблизительно 0 С;(б) нагревание реакции до приблизительно 75 С и поддержание данной температуры реакции в течение 5 ч или до тех пор, пока циклизация, по существу, не завершится;(в) охлаждение реакционной смеси до приблизительно 0 С;(г) гашение реакции насыщенным NH4Cl; и, необязательно,(д) очистка желаемого продукта циклопропилацетилена. Способ по настоящему изобретению представляет собой процесс, проходящий в одной емкости, который начинается смешиванием одного эквивалента 5-галоген-1-пентина в апротонном растворителе, по крайней мере, с приблизительно 1,0 эквивалентом сильного основания в апротонном растворителе при температуре в интервале между приблизительно -20 и приблизительно 150 С. Предпочтительный интервал эквивалентов сильного основания лежит между приблизительно 2,0 и приблизительно 2,5 эквивалентами. Предпочтительным исходным продуктом является 5-хлор-1-пентин. Предпочтительная температура для данного смешивания лежит в пределах интервала между приблизительно -20 и приблизительно 25 С, наиболее предпочтительно 0 С. Перед смешиванием апротонный растворитель для 5-галоген-1-пентина 4 может быть тем же или отличаться от апротонного растворителя для сильного основания. Сильное основание выбирают из группы,содержащей н-бутиллитий, амид натрия, диэтиламид натрия, гидрид натрия, гидрид калия, бис(триметилсилил) амид натрия, бис(триметилсилил) амид калия, ДАЛ, втор-бутиллитий, третбутиллитий и тетраметилпиперидид лития. Предпочтительным сильным основанием является н-бутиллитий. Апротонный растворитель выбирают из ТГФ, 2,5-диметилТГФ, 1,4-диоксана, диэтоксиметана, диметоксиэтана, циклогексана, гексана и гексана с тетраметилендиамином. Предпочтительным апротонным растворителем является циклогексан. Смешивание сильного основания и 5 галоген-1-пентина является экзотермической реакцией, приводящей к циклизации. Циклизация происходит спонтанно. Предпочтительно нагревать реакцию в мере, достаточной для ускорения циклизации. Предпочтительная температура для циклизации лежит в интервале между приблизительно 50 и приблизительно 80 С,предпочтительно около 75 С. Чем выше температура, тем короче время инкубации, необходимое для существенно полной циклизации. В случае температуры 75 С для завершения циклизации обычно требуется время инкубации около 5 ч. Понятно, что специалист в данной области легко определит вариации температуры и времени инкубации на данной стадии циклизации. Когда циклизация, по существу, завершена или завершена, по крайней мере, в достаточной мере, реакционную смесь можно, но необязательно охладить до температуры от приблизительно -30 до приблизительно 50 С, предпочтительно до температуры около 0 С. После этого в целях гашения реакции добавляют источник протонов. В данном изобретении источник протонов выбирают из насыщенного NH4Cl, HCl и Н 2SO4. Предпочтительным источником протонов является NH4Cl. В заключение, на данном этапе может быть включена стадия очистки для выделения циклопропилацетилена. Реакция, применяемая для введения циклопропилацетиленовых групп в структуры других молекул, как правило, имеет известный химизм и хорошо известна специалистам в данной области. Для введения циклопропилацетиленовой группы в ароматические заместители легко осуществляется катализируемое палладием связывание. Для введения циклопропилацетиленой группы в алкиловые заместители проводят реакцию замещения. Соединения по настоящему изобретению могут иметь в своем составе асимметрические центры и, за исключением тех случаев, когда это оговаривается особо, могут существовать в виде рацематов, рацемических смесей или от 5 дельных диастереомеров или энантиомеров,причем все изомерные формы включены в настоящее изобретение. Подразумевается, что термин "(+/-)" охватывает (+)-оптические изомеры или (-)-оптические изомеры или их смеси. Когда любая переменная (например, R, апротонный растворитель) появляется на любой стадии более одного раза, ее значение при каждом появлении является независимым от ее значения при каждом другом появлении. Также,сочетания заместителей и/или переменных разрешены только в том случае, если такие сочетания приводят к образованию устойчивых соединений. Подразумевается, что термин "алкил", такой как применяется здесь, за исключением тех случаев, когда это оговаривается особо, включает в себя насыщенные алифатические углеводородные группы как с разветвленной, так и с неразветвленной цепью, содержащие установленное количество атомов углерода. Термин "галоген" или "галоген-", такой как применяется здесь, означает фтор, хлор, бром и иод. В данном изобретении циклопропилацетилен получают по следующей схеме. Схема 1 Суммарный выход составляет свыше 65%. В противоположность этому, предшествующий способ с применением коррозивных реагентов дает выход около 42% и излагается ниже. Схема 2 как описывается также в L.E.Hudson et al., J.et al., Synthesis, 703 (1972). Соединение А может быть синтезировано по следующей методике. 7 Соединение А может использоваться для получения и проведения скрининговых исследований противовирусных соединений. Например, соединение А может использоваться для выделения ферментных мутантов, которые являются превосходными инструментами скрининга для наиболее мощных противовирусных соединений. Кроме того, соединение А может использоваться для установления и определения участка связывания других противовирусных средств на обратной транскриптазе ВИЧ, например, с помощью конкурентного ингибирования. Таким образом, соединение А является коммерческим продуктом, предназначенным для продажи в данных целях. Соединение А может использоваться для ингибирования обратной транскриптазы ВИЧ,предотвращения или лечения инфекции вируса иммунодефицита человека (ВИЧ) и лечения последующих патологических состояний, таких как СПИД. Лечение СПИД или предотвращение или лечение ВИЧ-инфекции определяется как включающее, но не ограничивающееся лечение широкого спектра состояний при ВИЧинфекции: СПИД, САК (СПИД-ассоциированного комплекса) как проявившегося, так и бессимптомного, и имевшего место или потенциального контакта с ВИЧ. Например, соединение по данному изобретению может использоваться для лечения ВИЧ-инфекции после имевшего место подозреваемого контакта с ВИЧ посредством, например, переливания крови, обмена жидкостей организма, укусов, случайного укола иглой или контакта с кровью пациента во время хирургической операции. Особенным преимуществом соединения А является его мощный ингибирующий эффект на обратную транскриптазу ВИЧ, ставшую устойчивой к другим противовирусным средствам,таким как L-697661, которое представляет собой 3-([(4,7-дихлор-1,3-бензоксазол-2-ил)метил] амино)-5-этил-6-метилпиридин-2(1 Н)-он; или L696229, которое представляет собой 3-[2-(1,3 бензоксазол-2-ил)этил]-5-этил-6-метилпиридин 2 (1 Н) -он; или AЗТ. В данных целях соединение А может вводиться перорально, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или инфузионные методы) , с помощью аэрозоля для ингаляций или ректально в составах стандартных доз, содержащих традиционные нетоксичные фармацевтически приемлемые носители,адъюванты и наполнители. Таким образом, в соответствии с настоящим изобретением описывается способ лечения и фармацевтическая композиция для лечения ВИЧ-инфекции и СПИД. Лечение включает в себя введение пациенту, нуждающемуся в таком лечении, фармацевтических композиций, содержащих фармацевтический носитель и тера 000074 8 певтически эффективное количество соединения по настоящему изобретению. Данные фармацевтические композиции могут быть в форме суспензий или таблеток для перорального применения; назальных аэрозолей; стерильных препаратов для инъекций, например, в форме стерильных водных или масляных суспензий для инъекций или суппозиториев. Если препарат вводят перорально в форме суспензии, данные композиции готовятся в соответствии с технологиями, хорошо известными в области технологий приготовления лекарственных средств, и могут содержать микрокристаллическую целлюлозу для придания большего объема, альгиновую кислоту или альгинат натрия в качестве суспендирующего средства,метилцеллюлозу в качестве усилителя вязкости и подсластители/ароматизаторы, известные в данной области. Будучи в виде таблеток немедленного высвобождения, данные композиции могут содержать микрокристаллическую целлюлозу, фосфат дикальция, крахмал, стеарат магния и лактозу и/или другие наполнители,связывающие вещества, сухие разбавители, разрыхлители, разбавители и смазывающие вещества, известные в данной области. Если препарат вводят в виде назального аэрозоля или ингаляций, данные композиции готовятся в соответствии с технологиями, хорошо известными в области технологий приготовления лекарственных средств, и могут быть приготовлены в виде растворов в физиологическом растворе с применением бензилового спирта или других подходящих консервантов,ускорителей всасывания для улучшения биологической доступности, фторуглеродов и/или других солюбилизирующих или диспергирующих средств, известных в данной области. Растворы или суспензии для инъекций могут быть приготовлены в соответствии с известными технологиями, применяя подходящие нетоксичные, приемлемые для парентерального введения, разбавители или растворители, такие как маннитол, 1,3-бутандиол, вода, раствор Рингера или изотонический раствор хлорида натрия или подходящие диспергирующие или увлажняющие и суспендирующие средства, такие как стерильные, мягкие, нелетучие масла, включая синтетические моно- и диглицериды, и жирные кислоты, включая олеиновую кислоту. Если препарат вводят ректально в форме суппозиториев, данные композиции могут быть приготовлены путем смешивания лекарственного средства с подходящим нераздражающим наполнителем, таким как масло какао, синтетические сложные эфиры глицеридов или полиэтиленгликоли, которые являются твердыми при обычных температурах, но плавятся и/или растворяются в полости прямой кишки с высвобождением лекарственного средства. Соединение А может вводиться людям перорально в интервале дозировок от 1 до 1000 мг/кг массы тела в разделенных дозах. Одним предпочтительным интервалом дозировок является от 0,1 до 10 мг/кг массы тела перорально в разделенных дозах. Другим предпочтительным интервалом дозировок является от 0,1 до 20 мг/кг массы тела перорально в разделенных дозах. Для комбинированной терапии с нуклеозидными аналогами предпочтительным интервалом дозировок является от 0,1 до 20 мг/кг массы тела для соединений по данному изобретению, вводимых перорально в разделенных дозах, и от 50 мг до 5 г/кг массы тела для нуклеозидных аналогов, вводимых перорально в разделенных дозах. Однако следует учитывать,что индивидуальный размер дозы и частота дозировки для каждого отдельного пациента может варьироваться и будет зависеть от ряда факторов, включая активность конкретного применяемого соединения, метаболическую устойчивость и продолжительность действия данного соединения, возраста, массы тела, общего состояния здоровья, пола, диеты, способа и времени введения, скорости выведения, комбинации лекарственных средств, тяжести конкретного состояния и терапии, которой подвергается данный пациент. Пример 1. Получение циклопропилацетилена. К раствору 5-хлор-1-пентина в циклогексане (80 мл) при 0 С в атмосфере N2 добавляли н-бутиллитий в циклогексане (2,0 М, 122 мл). Смесь нагревали до 75 С в течение 5 ч. Добавление н-бутиллития к алкину являлось экзотермическим, температуру во время данных добавлений поддерживали ниже +5 С,применяя баню лед-Н 2 О. Ход стадии циклизации отслеживали с помощью ЖХВР. Реакцию считали завершенной,когда анализируемый выход составлял 90%. Условия ЖХВР: колонка Phenyl, СН 3 СN,вода, фосфорная кислота; 50:50:0,1 изократическая элюция в течение 20 мин, скорость элю 10 ции=1,0 мл/мин, анализ в ультрафиолетовой области при 195 нм, tR исходного вещества=7,5 мин, tR циклопропилацетилена=6,0 мин. Продукт обладает фактором чувствительности в 20 раз большим, чем исходный продукт. Как только стадия циклизации завершалась, реакцию охлаждали до 0 С и гасили насыщенным NH4Cl. Анализ органической фазы с помощью ЖХВР показал наличие 5,5 г циклопропилацетилена (выход 85%). Продукт очищали путем фракционной перегонки через 6" х 0,5" колонку, набитую 4 мм стеклянной дробью. Отбирали фракцию с точкой кипения между 45-75 С. Это позволяло получить 4,2 г (65%) циклопропилацетилена в виде бесцветного масла. Пример 2. Получение 4-хлорфенилпиваламида. Продукты Количество мМоль MM 4-Хлоранилин 76 г 596 127,57 Пивалоилхлорид 74 мл 600 120,58(600 мл) добавляли насыщенный Na2 СО 3 (95 мл). Партию охлаждали до 10 С и в течение 45 мин по каплям добавляли пивалоилхлорид (74 мл). Партию перемешивали при 5-10 С в течение 60 мин, в то время как ход реакции отслеживался с помощью ЖХВР. Добавление пивалоилхлорида к анилину было экзотермическим. Условия ЖХВР: колонка С-8, СН 3 СN, вода, фосфорная кислота; элюция по градиенту от 40:60:0,1 до 80:20:0,1 в течение 20 мин, скорость элюции=1,0 мл/мин,анализ в ультрафиолетовой области при 245 нм,tR исходного вещества=7,2 мин, tR пиваламида=12,6 мин. Продукт выделяли путем фильтрования и промывали Д.И. водой (3 х 75 мл) и сушили в воздушной сушилке с отсосом в течение 10 мин. Продукт сушили в вакуумной печи при 40 С с продувкой N2 в течение 16 ч с получением 108,5 г продукта в виде мелкодисперсных белых игл Продукты Количество мМоль MM 4-Хлорфенилпивала- 10 г 47,2 211,69 мид(75 мл) в 500 мл 3-хгорлой колбе и смесь охлаждали до 0 С. К данному раствору по каплям добавляли H-BuLi/гексан (2,5 М, 38 мл), тогда как температуре внутренней среды давали подняться до +15 С. Партию выдерживали при 0 С в течение 2 ч. Добавление первого эквивалента н-BuLi к пиваламиду было чрезвычайно экзотермичным. Экзотермичность контролировали с помощью скорости добавления. К полученной светло-желтой суспензии добавляли чистый этилтрифторацетат (6,7 мл),тогда как температуре внутренней среды позволяли подняться до +10 С. Ход реакции отслеживали с помощью ЖХВР. Условия ЖХВР: колонка С-8, СН 3 СN, вода, фосфорная кислота; элюция по градиенту от 40:60:0,1 до 80:20:0,1 в течение 20 мин, скорость элюции=1,0 мл/мин, анализ в ультрафиолетовой области при 245 нм, tR исходного пиваламида=12,6 мин, tR-кетопиваламида=11,6 мин. Обычно отмечалось 85 А% продукта и 10-15 А% непрореагировавшего пиваламида. Реакцию гасили путем добавления 6 Н НС 1(10 мл) и Д.И. воды (20 мл). Анализ ЖХВР на данном этапе показывал наличие 13,1 г (90%) продукта. Раствор концентрировали до приблизительно 50 мл в вакууме и промывали этанолом(50 мл) в целях удаления гексана и ТГФ. К партии добавляли 6 Н НСl (40 мл) и смесь нагревали до кипения (80 С) в течение 1 ч. Анализ ЖХВР показывает наличие 8590 А% кетоанилина, 10 А% непрореагировавшего пиваламида. Таким образом, ацилированный 12 продукт претерпевает гидролиз, тогда как непрореагировавший пиваламид не вступает в реакцию. Анализируемый выход на данном этапе составлял 7,78 г (74%). Партию концентрировали до приблизительно 50 мл в вакууме, когда образовывался осадок (по-видимому, НС 1 соль продукта). После выдерживания в течение 1 ч партию фильтровали и промывали гексаном (3 х 30 мл). Промывания гексаном удаляют непрореагировавший пиваламид из продукта. Твердое вещество анализируют с помощью ЖХВР, чтобы убедиться в том, что он полностью удален на данном этапе. Фильтрат и смывы обычно содержат 1,2-1,5 г продукта (8-12%). Большая часть потерь продукта находилась в водном фильтрате. Соль сушили в вакуумной печи при 40 С в течение 16 ч с получением 10,4 г твердого вещества, чистота которого составляла 71,4 мас.%(260 мл) и нейтрализовали до рН около 6-7 с помощью 2 н. NaOH (15 мл). Обязательно не доводить рН до значений выше 9,0, поскольку в таком случае продукт разлагается. Полученное ярко-желтое твердое вещество выделяли путем фильтрования и промывали Д.И. водой (2 х 25 мл). Продукт сушили в вакуумной печи при 40 С в течение 16 ч с получением 6 г кетоанилина, чистота которого составляла 96,6 мас.% (выход 54%). Продукт подвергают дальнейшей очистке путем перекристаллизации из гексана. Пример 4. Получение N-4-метоксибензилкетоанилина. Продукты Кетоанилин п-Метоксибензилхлорид 4 Молекулярные сита Толуол Ацетон Гексан В 250 мл колбу помещали кетоанилин(50 г) и толуол (75 мл). Смесь перемешивали при 23 С в атмосфере N2 в течение 24 ч. Анализ методом ЖХВР показал, что соотношение в смеси продукта и исходного вещества составляло 1:1. Условия ЖХВР: колонка С-8, СН 3 СN, вода, фосфорная кислота; изократическая элюция при 65:35:0,1 в течение 20 мин, скорость элюции=1,0 мл/мин, анализ в ультрафиолетовой области при 260 нм, tR толуола=5,7 мин, tR исходного кетоанилина=6,5 мин, tR продукта=15,0 мин. Обычно отмечалось 25 А% толуола. В реакцию помещали свежие молекулярные сита (40 г) и перемешивали в течение дополнительных 3 дней при 23 С. Реакцию считали завершенной,когда оставалось менее чем 2 А% исходного вещества. Смесь фильтровали через целит и промывали ацетоном (7 х 75 мл), пока из целита не была вымыта большая часть желтизны. Фильтрат концентрировали с получением 27 г желтооранжевого масла, которое затвердевало при стоянии. Твердое вещество очищали путем его растворения в горячем гексане (100 мл). Партию охлаждали до комнатной температуры, затем до 0 С на бане лед-Н 2 О. После выдерживания в течение 1,5 ч продукт фильтровали и промывали холодным гексаном (2 х 10 мл). Продукт сушили на воздухе под тягой в течение 10 мин,затем в вакуумной печи при 40 С в течение 2 ч. Это позволяло получить 20,5 г (86%) яркожелтого порошка. Пример 5. Получение аминоспирта. 14 Пирролидинилэфедрин (264 мг) растворяли в ТГФ (2 мл) и смесь охлаждали до -5 С. К смеси в атмосфере N2 при -5 С по каплям добавляли чистый циклопропилацетилен (0,11 мл) и втор-бутиллитий (2,0 мл). Смесь выдерживали при -5 С, в течение 30 мин, затем охлаждали до-45 С. Добавление втор-бутиллития вызывало выделение тепла, причем температуру поддерживали между -5 и 0 С путем контроля скорости добавления. Кетон (175 мг) растворяли в ТГФ (1,0 мл) в атмосфере N2 и добавляли к анионной смеси в течение 2-3 мин, позволяя в процессе добавления температуре внутренней среды подняться до -40 С. Полученный светло-оранжевый раствор выдерживали при -40 С в течение 60 мин и гасили путем добавления 1 М лимонной кислоты(3 мл) и этилацетата (3 мл). Реакцию нагревали до температуры окружающей среды и слои разделяли. Нижний водный слой экстрагировали добавлением этилацетата (3 мл). Объединенные органические слои промывали 1 М лимонной кислотой (2 х 3 мл). Реакционную смесь анализировали методом ЖХВР на предмет процента превращения и ЭИ продукта. Условия ЖХВР: колонка С-8,СН 3 СН:вода:фосфорная кислота, изократическая элюция 65:35:0,1 в течение 20 мин, скорость элюции=1,0 мл/мин, анализ в ультрафиолетовой области при 252 нм, tR исх. вещества=12,8 мин, tR продукта=10,3 мин. Условия хиральной ЖХВР: колонка с амилозой в качестве неподвижной фазы, изократическая элюция гексаном:изопропанолом 85:15,скорость элюции=1,0 мл/мин, анализ в ультрафиолетовой области при 252 нм, tR исх. вещества=4,9 мин, tR основного энантиомера=5,5 мин,tR минорного энантиомера=25,0 мин. Энантиомерный избыток составлял 98%, а процент химического превращения составлял 93%, (6 А% исходного вещества). Анализируемый выход составлял 92%. Пример 6. Получение бензоксазинона. 15 Продукты Аминоспирт Фосген в толуолеEtOAc ГГксаны 1 М лимонная кислота Насыщенный солевой раствор Аминоспирт растворяли в ТГФ (15 мл) и охлаждали до -10 С в атмосфере N2. К смеси добавляли триэтиламин (5,4 мл) и фосген в толуоле (4,6 мл). Добавление фосгена вызывало выделение тепла, причем температуру поддерживали ниже 20 С путем контроля скорости добавления. Ход реакции отслеживали с помощью ЖХВР, и она обычно завершалась за 15 мин. Условия ЖХВР: колонка С-8, СН 3 СN: вода: фосфорная кислота, элюция по градиенту от 50:50:0,1 до 90:10:0,1 в течение 20 мин, скорость элюции=1,5 мл/мин, анализ в ультрафиолетовой области при 252 нм, tR исх. продукта=14,6 мин,tR продукта =16,0 мин. Реакцию охлаждали до 0 С и гасили добавлением льда - холодной воды (15 мл) и этилацетата (20 мл). Насыщенный солевой раствор применяли для диспергирования любых эмульсий. Органический слой удаляли, а водный экстрагировали добавлением этилацетата (15 мл). Объединенные органические слои промывали 1 М лимонной кислотой (40 мл) и насыщенным солевым раствором (25 мл). Органический слой сушили (Na2SO4) и концентрировали в вакууме с получением 3,8 г коричневого масла. Продукт кристаллизовали из 5:1 гексана:этилацетата (25 мл), охлаждали до 0 С, выдерживали в течение 1 ч и фильтровали. Осадок с фильтра промывали холодным 5:1 гексаном:этилацетатом (2 х 5 мл). Осадок сушили на воздухе под тягой с получением 2,9 г (85%) светло-оранжевого твердого вещества. Пример 7. Получение соединения А. 16 Продукты ПМБ соединение А,7 Аммоний-нитрат церияCH3CN Этилацетат Д.И. вода Насыщенный солевой раствор Количество мМоль ММ 0,8 г 1,83 435 4,4 г 8,0 548,23 15 мл 30 мл 30 мл 10 мл Соединение А, защищенное п-метоксибензилом, растворяли в СН 3 СN (15 мл). К данному раствору добавляли раствор аммоний-нитрата церия (4,4 г) в воде (5 мл). Реакцию обычно завершали за 2 ч при 23 С, как было определено с помощью ЖХВР. Условия ЖХВР: колонка С-8, СН 3 СN: вода: фосфорная кислота, элюция по градиенту от 50:50:0,1 до 90:10:0,1 в течение 20 мин, скорость элюции=1,5 мл/мин, анализ в ультрафиолетовой области при 252 нм, tR исх. продукта=16,0 мин,tR продукта=9,0 мин. Реакцию разбавляли Д.И. водой (5 мл) и концентрировали приблизительно до 1/2 объема. Продукт экстрагировали из полученного водного слоя добавлением этилацетата (2 х 15 мл). Объединенные органические слои промывали Д.И. водой (2 х 10 мл) и солевым раствором (10 мл). Органику концентрировали в вакууме с получением желтого клейкого вещества. Продукт выделяли путем хроматографии на силикагеле. Пример 8. N-(4-хлорфенил)-2,2-диметилпропанамид. В 5 л 3-хгорлую круглодонную колбу с воздушной мешалкой добавляли 4-хлоранилин(127,57 г, 1 моль), 1200 мл СНСl3 и 1200 мл насыщенного водного раствора На 2 СО 3. К колбе прикрепляли капельную воронку и вносили 2,2 диметилпропаноилхлорид (129 мл, 1,05 моль). К энергично перемешиваемой смеси по каплям в течение 1 ч добавляли хлорангидрид. Полученную смесь перемешивали при температуре окружающей среды в течение дополнительных 23 ч. Некоторое количество продукта выделялось из смеси в виде белых кристаллов. Данные кристаллы отбирали путем фильтрования. Фильтрат переносили в разделительную воронку и слои разделяли. Хлороформный слой промывали водой и солевым раствором. Сушка (МgSO4),фильтрование и удаление растворителя в вакууме давали дополнительный продукт. Две порции продукта объединяли и перекристаллизовывали из кипящих EtOAc-гексанов с получением 185,6 г N-(4-хлорфенил)-2,2-диметилпропанамида в виде белого кристаллического твердого вещества. Пример 9. (-)-6-Хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2 Н-3,1-бензоксазин-2-он (соединение А) и (+)-6-хлор-4 циклопропилэтинил-4-трифторметил-1,4-дигидро-2 Н-3,1-бензоксазин-2-он. 17 Стадия А. 2-(2-Амино-5-хлорфенил)-4 циклопропил-1,1,1-тpифтop-3-бутин-2-oл. Раствор циклопропилацетилида броммагния готовили из 23 г циклопропилацетилена(0,348 моль) в 250 мл ТГФ путем добавления по каплям 116 мл 3,0 М раствора бромида этилмагния в эфире (0,348 моль) в течение 1 ч. Температуру данного раствора поддерживали на 0 С в течение 1 ч, затем на 40 С в течение 3 ч. К данному раствору, вновь охлажденному до 0 С,добавляли 15,56 г 1-(2-амино-5-хлорфенил)2,2,2-трифторметилэтанона (0,0696 моль) в виде твердого вещества порциями через каждые 5 мин. Реакционной смеси давали перемешиваться при 0 С в течение 1,5 ч. Реакцию гасили при 0 С путем капельного добавления 700 мл насыщенного водного раствора хлорида аммония. Смесь экстрагировали добавлением 2 х 400 мл порций этилацетата, объединенные органические фазы промывали солевым раствором и сушили над MgSO4. Удаление осушителя и растворителей давало желтое твердое вещество. Данный продукт перекристаллизовывали из кипящих гексанов (конечный объем 100 мл) с получением 14,67 г 2-(2-амино-5-хлорфенил)-4 циклопропил-1,1,1-трифтор-3-бутин-2-ола. Второй сбор (2,1 г) получали путем концентрирования маточных растворов. т. пл.: 153-154 С. 1 НЯМР (CDCl3):0,84 (м, 2 Н), 0,90 (м, 2 Н), 1,38(15,00 г, 0,0518 моль) и 41,98 г (0,259 моль) 1,1'карбонилдиимидазола в 250 мл сухого ТГФ перемешивали в атмосфере аргона при 55 С в течение 24 ч. Растворитель удаляли с помощью роторного испарителя и остаток делили между 500 мл этилацетата и 400 мл воды. Слои разделяли и водную фазу экстрагировали добавлением этилацетата еще раз. Объединенные этилацетатные экстракты промывали 2 х 200 мл 2% водного НС 1, насыщенным водным NаНСО 3 и солевым раствором. Сушка над МgSO4, фильтрование и удаление растворителя в вакууме давали 16,42 г соединения заголовка в виде твердого вещества. Перекристаллизация из этилацетата-гексана позволяла получить 12,97 г аналитически чистого -6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2 Н-3,1-бензоксазин-2-она в виде белых кристаллов, т.пл. 178-180 С. 1 Н-ЯМР (СОС 13):0,85 (м, 2 Н), 0,94 (м,2 Н), 1,40 (м, 1 Н), 6,81. (д, J=8,5 Гц, 1 Н), 7,37 (дд,J=2,5, 8,5 Гц, 1 Н), 7,49 (д, J=2,5 Гц, 1 Н), 8,87(14,22 г, 0,06556 моль) в 350 мл сухого дихлорметана, перемешиваемому в атмосфере аргона на ледяной бане, добавляли триэтиламин (22,84 мл, 0,164 моль). Охлаждающую баню удаляли и реакции давали протекать при комнатной температуре. Спустя 75 мин реакцию считали завершенной на основании данных тонкослойной хроматографии (SiO2, 4% EtOAc в СНСl3) и раствор разбавляли 500 мл СНСl3, затем промывали 10% лимонной кислотой (2 Х), водой (1X) и солевым раствором (1X). Сушка (MgSO4), фильтрование и удаление растворителя в вакууме давали бесцветную пену. Данный продукт растирали с 200 мл кипящего гексана. При охлаждении до комнатной температуры желаемый диастереомерный камфанатимид выпадал в осадок. Твердое вещество собирали на фритту, промывали небольшим количеством холодных гексанов и сушили в вакууме с получением 7,79 г 6 хлор-1-(1S)-камфаноил-4-циклопропилэтинил 4-трифторметил-1,4-дигидро-2 Н-3,1-бензоксазин-2-она в виде белых кристаллов, т. пл.: 164165 С. Чистота по данным ЖХВР: 99,2%254 нм. 1 Н-ЯМР (CDCl3):0,77 (с,3 Н), 0,86-0,96 (м,4 Н), 1,08 (с, 3 Н), 1,19 (с, 3 Н), 1,44 (м, 1 Н), 1,76(м, 1 Н), 1,95 (м, 1 Н), 2,15 (м, 2 Н), 7,42 (дд, J=2,4,9,0 Гц, 1 Н), 7,63 (м, 2 Н). Стадия Г. (-)-6-Хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2 Н-3,1-бензоксазин-2-он (соединение А). 6-Хлор-1-(1S)-камфаноил-4-циклопропилэтинил-4-трифторметил-1,2-дигидро-4(Н)-3,1 бензоксазин-2-он (7,50 г, 0,01512 моль) растворяли в 150 мл н-бутанола при 60 С в атмосфере аргона. К данному раствору добавляли 10 мл 1 н. НСl. Данный раствор вьдерживали при 60 С в течение 72 ч. Смесь нейтрализовали воднымNaHCO3 и н-бутанол удаляли в вакууме. Остаток растворяли в 150 мл ТГФ и обрабатывали 50 мл 2 н. LiOH в течение 3 ч при комнатной температуре. Данную смесь разбавляли этилацетатом и промывали двумя порциями воды и одной солевого раствора. Сушка (MgSO4), фильтрование и удаление растворителя в вакууме давали белое твердое вещество. Данный продукт перекристаллизовывали из горячего гексана с получением 3,43 г (-)-6-хлор-4-циклопропилэтинил 4-трифторметил-1,4-лигидро-2 Н-3,1-бензоксазин-2-он в виде белых кристаллов. т.пл.: 131132 С; []D20=-84,7 (СНСl3, с=0,005 г мл-1); 1 НЯМР (СОСl3):0,85 (м, 2 Н), 0,94 (м, 2 Н), 1,40 19 Гц, 1 Н), 7,49 (д, J=2,5 Гц, 1 Н), 8,87 (ушир. с,1 Н). Стадия Д. (+)-6-Хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2 Н-3,1 бензоксазин-2-он. Маточные растворы из стадии В, описанной выше, очищали методом колоночной хроматографии на силикагеле, применяя 10% этилацетат в гексанах в качестве элюента. Чистый нежелательный диастереомер (бесцветную пену) гидролизовали в соответствии с методикой из стадии Г. Энантиомерный бензоксазинон, (+)(СDСl3):0,85 (м, 2 Н), 0,94 (м, 2 Н), 1,40 (м,1 Н), 6,81 (д, J=8,5 Гц, 1 Н), 7,37 (дд, J=2,5, 8,5 Гц, 1 Н), 7,49 (д, J=2,5 Гц, 1 Н), 8,87 (ушир. с,1 Н). Анализ обратной транскрептазы. В анализе изучали включение меченного тритием дезоксигуанозинтрифосфата рекомбинантной обратной транскриптазой ВИЧ (HIVRTR) (или другой RТ) в кислотно-осаждаемую кДНК при значениях КM dGTP и полиr(С)oлигo d(G)12-18. Ингибиторы, заявленные согласно настоящему изобретению, ингибируют такое включение. Исследования проводили в 55 мМ ТрисN,N,N',N'тетрауксусной кислоты (ЭГТУ), 1 мг бычьего сывороточного альбумина на мл. После 60 мин инкубации при 37 С кислотно-осаждаемый продукт собирали на фильтры со стекловолокном с помощью полуавтоматического харвестера клеток. Экстракты бактериальных клеток, содержащие RТ, разбавляли до тех пор, пока не достигали линейного интервала, и определяли активность в присутствии и в отсутствии ингибитора. Очищенный гетеродимер HIV-1 RT, продуцированный в Е. coli, также служил в качестве контроля. Результаты определяли как концентрацию ингибитора, дающую 50% ингибирование (КИ 50 маc.), в наномолях/литр. Соединение А давало KM50 маc. 2 нМ. Для проведения двойного мутантного анализа (дм) применяли A17RT. A17RT устойчива к различным аминопиридонам, как описано вNunberg, J.H. et al., J. Virol., 65, 4887 (1991). Результаты измеряют как KИ 50 дм в наномолях/литр. Соединение А давало КИ 50 мас. 85 нМ. Клеточный анализ распространения. Ингибирование распространения ВИЧ в культуре клеток измеряли по Nunberg, J.H. et al.,J. Virol., 65, 4887 (1991). В данном анализе Т 000074 20 лимфоидные клетки МТ-4 были инфицированы ВИЧ-1 (дикий тип, если только это не оговаривается особо), применяя заранее определенный инокулят и культуры инкубировали в течение 24 ч. В данный момент 1% клеток давали положительный результат по иммунофлюоресценцию. Клетки затем обильно промывали и распределяли в 96-луночные чашки для культивирования. В лунки добавляли последовательные двукратные разведения ингибитора растили в течение дополнительных 3 суток. Через 4 дня после внесения инфекции в контрольных культурах было инфицировано 100% клеток. Накопление HIV-1 р 24 напрямую коррелировало с распространением вируса. Ингибирующую концентрацию в клеточной культуре определяли как концентрацию ингибитора в наномолях/литр, которая сокращала распространение инфекции, по крайней мере, до 95%, или КИК 95. Несмотря на то, что описание, данное выше, знакомит с принципами настоящего изобретения вместе с примерами, приведенными в целях иллюстрации, следует учитывать, что практическое применение изобретения охватывает все обычные вариации, адаптации или модификации, которые попадают в объем следующей формулы изобретения и ее эквивалентов. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения циклопропилацетилена, включающий следующие стадии:(а) смешивание, по крайней мере, около 1,0 эквивалента сильного основания, выбранного из группы, включающей н-бутиллитий, амид натрия, диэтиламид натрия, гидрид натрия, гидрид калия,бис(триметилсилил)амид натрия,бис(триметилсилил)амид калия, диизопропиламид лития, втор-бутиллитий, трет-бутиллитий и тетраметилпиперидид лития, в апротонном растворителе с одним эквивалентом 5-галоген-1 пентина в апротонном растворителе при температуре в интервале между около -20 и около 150 С;(б) повышение температуры реакционной смеси до температуры от около 0 до около 150 С и поддержание температуры в пределах данного интервала, по крайней мере, в течение 15 мин или до тех пор, пока циклизация по существу не завершится; и(в) гашение реакции любым источником протонов. 2. Способ получения циклопропилацетилена, включающий следующие стадии: творителе с одним эквивалентом 5-галоген-1 пентина в апротонном растворителе при температуре в интервале между около -20 и около 150 С;(б) повышение температуры реакционной смеси до температуры от около 0 до около 150 С и поддержание температуры в пределах данного интервала, по крайней мере, в течение 15 мин или до тех пор, пока циклизация по существу не завершится;(в) охлаждение реакционной смеси до температуры в интервале между около -30 и около 50 С;(г) гашение реакции любым источником протонов. 3. Способ по п.2, дополнительно включающий очистку циклопропилацетилена. 4. Способ по п.1, где сильным основанием является н-бутиллитий. 5. Способ по любому из пп.1-3, где апротонный растворитель выбран из тетрагидрофурана, 2,5-диметилтетрагидрофурана, 1,4-диоксана, трет-бутилметилового эфира, диэтокси 22 метана, диметоксиэтана, циклогексана, гексана и смеси гексана с тетраметилендиамином. 6. Способ по п.5, где апротонным растворителем является циклогексан. 7. Способ по любому из пп.1-3, где источник протонов выбран из насыщенного NH4Cl,НСl и Н 2SO4. 8. Способ по любому из пп.1-3, где 5 галоген-1-пентином является 5-хлор-1-пентин. 9. Способ получения циклопропилацетилена, включающий следующие стадии:(а) смешивание от около 2,0 до около 2,5 эквивалентов н-бутиллития в циклогексане с одним эквивалентом 5-хлор-1-пентина в циклогексане при около 0 С;(б) нагревание смеси до температуры около 75 С и поддержание данной температуры в течение 5 ч или до тех пор, пока циклизация по существу не завершится;(в) охлаждение реакционной смеси до температуры около 0 С;(г) гашение реакции насыщенным NH4Cl. 10. Способ по п.9, дополнительно включающий очистку циклопропилацетилена.
МПК / Метки
МПК: C07C 13/04
Метки: получения, способ, циклопропилацетилена
Код ссылки
<a href="https://eas.patents.su/12-74-sposob-polucheniya-ciklopropilacetilena.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения циклопропилацетилена.</a>
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Ансель Жан-Эрик, Бьенейм Юг, Мейллян Пьер
МПК: A61K 31/355, B01J 31/24, C07C 39/19...
Метки: использованием, способ, замещенных, витамина, cпособ, фенолов, получения
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Термопластичный концентрат и способ его получения.

Номер патента: 49
Опубликовано: 30.04.1998
Автор: Стрэмел Родни
Метки: концентрат, термопластичный, способ, получения
Формула / Реферат:
1. Термопластичный концентрат, содержащий неорганический пигмент, диспергированный в термопластичной смоле, отличающийся тем, что пигмент содержит осажденный на нем агент для обработки, представляющий собой диалкилсульфосукцинат, соответствующий формуле ROOCCHSO3MCH2COOR', в которой R и R'обозначают одновалентные алкильные радикалы, содержащие от около 2 до около 20 атомов углерода, и М является катионом одновалентного металла; в...
Мезопористый алюмогель и способ его получения

Номер патента: 16
Опубликовано: 30.12.1997
Авторы: Беллусси Джузеппе, Миллини Роберто, Калемма Винченцо, Перателло Стефано
МПК: C04B 35/10, B01J 21/04, C01F 7/02...
Метки: мезопористый, способ, алюмогель, получения
Формула / Реферат:
1. Мезопористый гель, содержащий матрицу оксида алюминия, в которой могут быть гомогенно диспергированы один или более оксидов, выбранных из группы, включающей диоксид кремния, оксид бора, оксид фосфора, оксид металла группы VIII и/или VIB общей формулы МОx, при следующих молярных соотношениях между указанными оксидами и оксидом алюминия: SiO2/Al2O3 = 0-3,0 B2O3/Al2O3 = 0-4,0 P2O5/Al2O3 = 0-0,2 МОx/Аl2O3 = 0-0,2, и имеющий удельную...
Производные полипирролкарбоксамидонафталина, способ их получения и их применение

Номер патента: 6
Опубликовано: 30.12.1997
Авторы: Бьясоли Джиованни, Чомеи Марина, Монджелли Никола, Ломбарди Борджиа Андреа, Пезенци Энрико, Анджелуччи Францеско
МПК: C07H 15/252, A61K 31/40, C07D 207/34...
Метки: применение, способ, получения, производные, полипирролкарбоксамидонафталина
Формула / Реферат:
1. Соединение формулы (II): где R является кислотной группой; m - целое число от 1 до 3; n - ноль или целое число от 1 до 3; А представляет собой ферментативно гидролизуемый спейсер; Х является биологически активным соединением; или его фармацевтически приемлемые соли. 2. Соединение формулы (II) по п.1, где R является кислотной группой, выбранной из сульфоновой, карбоксильной и фосфоновой кислотных групп. 3. Соединение формулы (II)...
Способ получения микромезопористого геля

Номер патента: 13
Опубликовано: 30.12.1997
Авторы: Миллини Роберто, Беллусси Джузеппе, Перего Джованни, Паццукони Джаннино, Басси Джанлука, Перего Карло
МПК: C01B 33/46, B01J 20/18
Метки: микромезопористого, способ, геля, получения
Формула / Реферат:
1. Способ получения микромезопористого геля, содержащего силикагелевую матрицу с однородным распределением пор, в которой могут быть гомогенно распределены один или более оксидов металлов, выбранных из переходных металлов или из металлов групп IIIA, IVA и VA, включающий: а) проведение гидролиза и гелеобразования тетраалкилортосиликата, взятого в чистом виде или в спиртовом растворе, при температуре 20-80°С с водным раствором гидроксида...
Предыдущий патент: Способ повышения эффективности используемых при добыче нефти химикатов
Случайный патент: Рабочее колесо насоса центробежного или полуаксиального типа