Новые соединения бензоиндолина, способ их получения и фармацевтические композиции, которые их содержат
Номер патента: 7231
Опубликовано: 25.08.2006
Авторы: Миллан Марк, Гобер Ален, Де-Кара Бенжамен, Мюлле Оливье, Лавьелль Жильбер









Формула / Реферат
1. Соединения формулы (I)

в которой R1 и R2 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино, диалкиламино или группой трифторметила, a R3 и R4 представляют собой атом водорода, или
R1 и R4 представляют собой атом водорода, a R2 и R3 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино, диалкиламино или группой трифторметила, или
R1 и R2 представляют собой атом водорода, а R3 и R4 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино, диалкиламино или группой трифторметила,
их энантиомеры и диастереоизомеры, их аддитивные соли с фармацевтически приемлемой кислотой или основанием,
при этом понятно, что
термин "алкил" означает неразветвленную или разветвленную углеводородную цепь, содержащую от 1 до 6 атомов углерода,
термин "алкокси" означает неразветвленную или разветвленную группу алкилокси, содержащую от 1 до 6 атомов углерода.
2. Соединения формулы (I) в соответствии с п.1, в которых R1 и R2 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино, диалкиламино или группой трифторметила, a R3 и R4 представляют собой атом водорода, их энантиомеры и диастереоизомеры, а также их аддитивные соли с фармацевтически приемлемой кислотой или основанием.
3. Соединение формулы (I) в соответствии с п.1 или 2, которое представляет собой N-[4-метокси-3-(4-метил-1-пиперазинил)фенил]-1,2-дигидро-3H-бензо[е]индол-3-карбоксамид, а также его аддитивные соли с фармацевтически приемлемой кислотой или основанием.
4. Способ получения соединения формулы (I) в соответствии с п.1, отличающийся тем, что в качестве исходного соединения используют бензоиндольное соединение формулы (II)

в которой R1, R2, R3 и R4 являются такими, как определено для формулы (I), которое конденсируют с соединением формулы (III)

в которой Y представляют собой группу -N=C=O или -C(O)-N3, для получения соединения формулы (I),
которое может быть очищено, в случае необходимости, в соответствии с традиционным способом очистки,
которое разделяют на изомеры (диастереоизомеры и энантиомеры), в случае необходимости, в соответствии с традиционным способом разделения,
которое превращают, если это является желательным, в его аддитивные соли при использовании приемлемой кислоты или основания.
5. Фармацевтические композиции, включающие в качестве активного ингредиента по крайней мере одно соединение в соответствии с любым из пп.1-3, отдельно или в комбинации с одним или более инертными, нетоксическими, фармацевтически приемлемыми наполнителями или носителями.
6. Фармацевтические композиции в соответствии с п.5, включающие по крайней мере один активный ингредиент в соответствии с любым из пп.1-3, для применения в качестве лекарственного средства с двойственной a 2-AR/5-НТ2C антагонистической активностью.
7. Фармацевтические композиции в соответствии с п.5, включающие по крайней мере один активный ингредиент в соответствии с любым из пп.1-3, для применения при производстве лекарственных средств для лечения депрессии, беспокойства, расстройств поведения, связанных с повышенной возбудимостью, шизофрении, болезни Паркинсона, когнитивных расстройств, расстройств либидо и сексуальных дисфункций, а также расстройств сна.
Текст
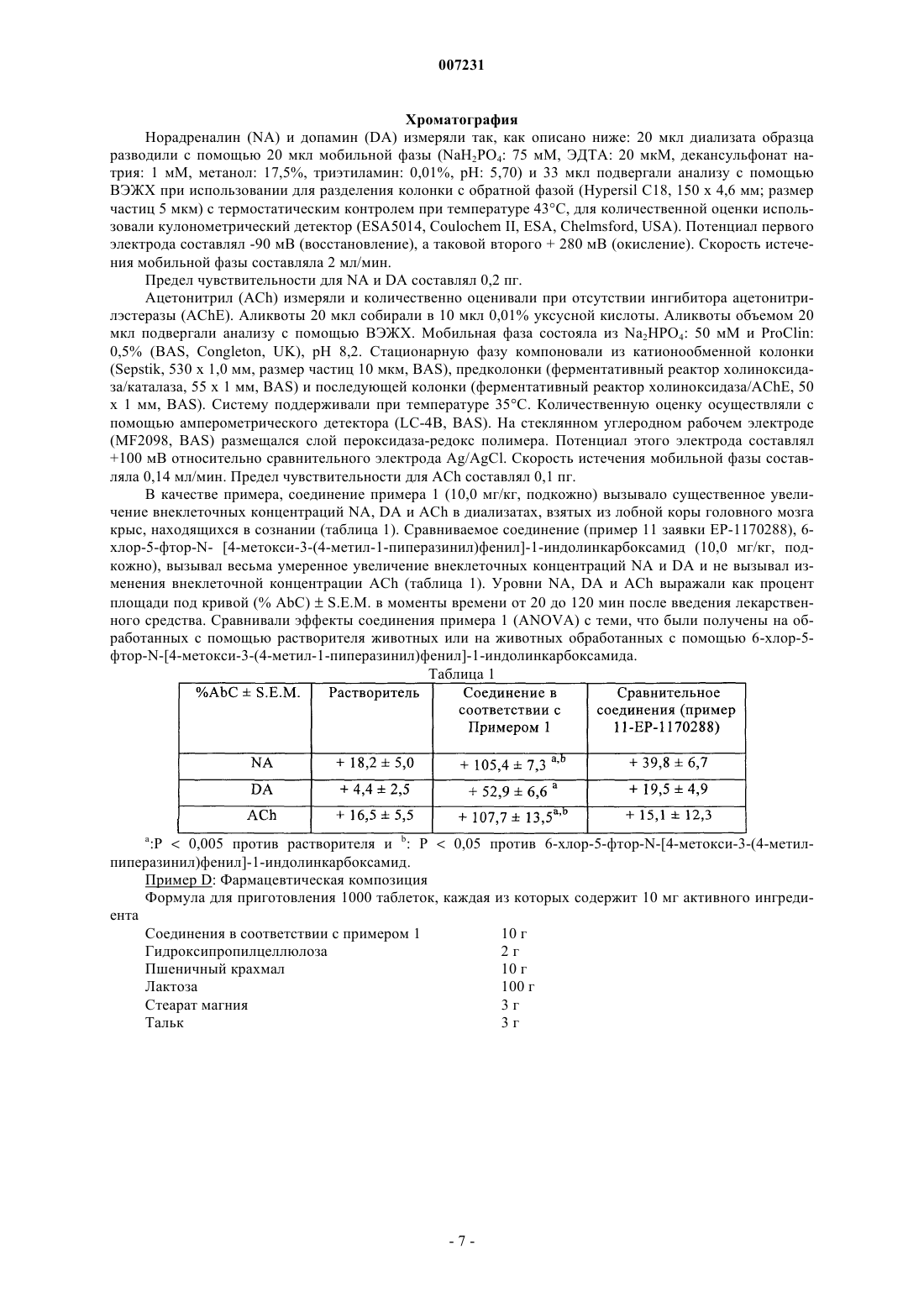
007231 Настоящее изобретение, вариант выбора касательно патента ЕР-1170288, относится к новым соединениям бензоиндолина, способу их получения и фармацевтической композиции, которая их содержит. Лобная часть коры головного мозга играет основную роль в процессах контроля измененных функций при психиатрических расстройствах. В частности, сейчас признается, что нарушение норадренергической, допаминергической и холинергической передачи в значительной степени вовлечено в этиологию различных расстройств такого рода. Например, в случае депрессии активность таких нейромедиаторов снижается в кортиколимбических участках. Среди различных классов ауто- и гетерорецепторов моноаминов, вовлеченных в механизмы регуляции, 2-AR (адренорецепторы) и 5-НТ 2 С рецепторы были определены как такие, которые обладают особой важностью. Два таких подтипа рецепторов действуют в одном направлении путем ингибирования допаминергической и адренергической трансмиссии. С одной стороны, ингибиторный контроль приводится в действие рецепторами 2-AR на допаминергических, норадренергических и холинергических нейронах [TheJournal of Pharmacology and Experimental Therapeutics, 270, 958 (1994); European Journal of Neuroscience,12, 1079 - 1095 (2000), The Journal of Pharmacology and Experimental Therapeutics, 305, 338 - 346 (2003)], а с другой стороны, рецепторы 5-НТ 2 С приводят в действие ингибиторный контроль при допаминергической и норадренергической трансмиссии [Neuropharmacology, 36, 609 (1997)]. Соединения, связывающиеся с одним или несколькими из таких классов рецепторов, были ранее продемонстрированы как такие, которые обладают потенциалом для лечения ряда патологий. Например, благоприятная роль соединений, обладающих антагонистической активностью в отношении 2-AR была изучена при лечении когнитивных нарушений [Psychopharmacology, 123, 239-249[British Journal of Pharmacology, 124, 1550-1556 (1998)] и депрессии [European Journal of Neuroscience, 12,1079-95 (2000); The Journal of Pharmacology and Experimental Therapeutics, 277 (2), 852 -60 (1997); NaunynSchmiedeberg's Archiv. Pharmacol, 355 (1), 20 - 9 (1997)]. Кроме того, соединения, проявляющие антагонистическую активность в отношении рецептора 5 НТ 2 С, продемонстрировали свою полезность при лечении сексуальных дисфункций [Life Sciences, 45,1263-1270 (1989); Pharmacological Biochemistry and Behavior, 39, 605-612 (1991); Psychopharmacology,119, 291-294 (1995); Neuroendocrinology, 76, 28-34 (2002)] болезни Паркинсона [Drug News Perspect, 12Neuropsychopharmacology, 21, 455-466 (1999); Biological Psychiatry, 47, 468-470 (2000); Pharmacology,Biochemistry and Behavior, 71, 599-605 (2002)]. Соединения, обладающие двойственным антагонистическим характером в отношении 2-AR и 5 НТ 2 С, могут быть чрезвычайно полезными для клиницистов в получении путем введения единственного соединения значительно усиленного действия с помощью эффекта синергизма при восстановлении нейротрансмиссии. Соединения такого рода имеет значительное преимущество перед введением двух различных соединений. Соединения в соответствии с изобретением обладают новой структурой бензоиндолина, которая обеспечивает им двойственный 2-AR/5-HT2C антагонистический характер, и, таким образом, они являются полезными при лечении депрессии, беспокойства, шизофрении, болезни Паркинсона, когнитивных расстройств, расстройств либидо и сексуальных дисфункций, расстройств сна и расстройств поведения,связанный с повышенной возбудимостью. Настоящее изобретение относится к новым соединениям бензоиндолина, которые составляют вариант по отношению к соединениям, описанным в заявке ЕР-1170288. Эти соединения являются новыми и отличаются от тех, что описаны и упомянуты как примеры в заявке ЕР-1170288, не только отсутствием атома галогена в индолине, но, в частности, наличием бензогруппы, приконденсированной к группе индолина. Неожиданно было обнаружено, что введение этой группы обеспечивает соединения в соответствии с изобретением с фармакологическими свойствами, которые значительно превосходят таковые для-1 007231 структурно ближайшего соединения уровня техники (пример 11 заявки ЕР-1170288: 6-хлор-5-фтор-N-[4 метокси-3-(4-метил-1-пиперазинил)фенил]-1-индолинкарбоксамид). Применение радикала бензоиндолина, соответственно, сделало возможным значительное усовершенствование фармакологических свойств соединений в соответствии с изобретением. В частности, настоящее изобретение относится к соединениям формулы (I) в которой R1 и R2 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино, диалкиламино или группой трифторметила, a R3 и R4 представляют собой атом водорода, илиR1 и R4 представляют собой атом водорода, a R2 и R3 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино,диалкиламино или группой трифторметила, илиR1 и R2 представляют собой атом водорода, a R3 и R4 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино,диалкиламино или группой трифторметила,к их энантиомерам и диастереоизомерам, к их аддитивным солям с фармацевтически приемлемой кислотой или основанием,при этом понятно, что термин "алкил" означает неразветвленную или разветвленную углеводородную цепь, содержащую от 1 до 6 атомов углерода,термин "алкокси" означает неразветвленную или разветвленную группу алкилокси, содержащую от 1 до 6 атомов углерода. Среди фармацевтически приемлемых солей могут быть упомянуты хлористо-водородная кислота,бромисто-водородная кислота, серная кислота, фосфоновая кислота, уксусная кислота, трифторуксусная кислота, молочная кислота, пировиноградная кислота, малоновая кислота, янтарная кислота, глютаровая кислота, фумаровая кислота, винная кислота, малеиновая кислота, лимонная кислота, аскорбиновая кислота, метансульфоновая кислота, камфарная кислота, и т. д. Среди фармацевтически приемлемых основания могут быть упомянуты гидроокись натрия, гидроокись калия, триэтиламин, трет-бутиламин, и т. д. Предпочтительный аспект изобретения относится к соединениям, в которыхR1 и R2 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино, диалкиламино или группой трифторметила, aR3 и R4 представляют собой атом водорода. Среди предпочтительных соединений в соответствии с изобретением может быть, в частности, упомянут,N-[4-метокси-3-(4-метил-1-пиперазинил)фенил]-1,2-дигидро-3H-бензо[е]-индол-3-карбоксамид. Настоящее изобретение относится также к способу получения соединений формулы (I), характеризующемуся тем, что в качестве исходного материала используют соединение бензоиндола формулы (II) в которой R1, R2, R3 и R4 являются такими, как определено для формулы (I), которое конденсируют с соединением формулы (III) в которой Y представляет собой группу -N=C=O или -C(O)-N3, для получения соединения формулы (I),-2 007231 которое может быть очищено, в случае необходимости, в соответствии с традиционным способом очистки,которое разделяют на изомеры (диастереоизомеры и энантиомеры), в случае необходимости, в соответствии с традиционным способом разделения,которое превращают, если это является желательным, в его аддитивные соли при использовании приемлемой кислоты или основания,при этом понятно, что индолин формулы (II) получают в соответствии с известными способами,например, начиная с соответствующего соединения нитронафтилацетонитрила. Настоящее изобретение относится к фармацевтическим композициям, содержащим в качестве активного ингредиента по крайней мере одно соединение формулы (I), в отдельности или в комбинации с одним или более инертными, нетоксическими, фармацевтически приемлемыми наполнителями или носителями. Среди фармацевтических композиций в соответствии с изобретением могут быть упомянуты более подробно те, которые являются приемлемыми для перорального, парентерального, назального или трансдермального введения, таблеток или драже, подъязычных таблеток, желатиновых капсул, таблеток продолговатой формы, суппозиториев, кремов, мазей, дермальных гелей, и т. п. Полезные дозы варьируют в зависимости от возраста и веса тела пациента, природы и тяжести расстройства, а также от способа введения, который может быть пероральным, назальным, ректальным или парентеральным. Единица дозы обычно колеблется в пределах от 0,05 до 500 мг в течение 24 ч при количестве введений от одного до трех. Примеры, которые приведены ниже, иллюстрируют изобретения без ограничения его любым способом. Структуры соединений, которые описаны, были подтверждены при использовании традиционных способов спектроскопии и спектрометрии. Используемые исходные материалы представляют собой известные продукты или их готовят в соответствии с известными способами. Приготовление 1: 2,3-дигидро-1H-бензо[е]индол Этап А: (2-нитро-1-нафтил)ацетонитрил Готовили раствор 53,5 г (0,477 моля) трет-бутанолата калия в 400 мл диметилформамида. Охлаждали полученный раствор до температуры -10 С и прибавляли к нему в течение 1 ч раствор 40 г 4 хлорфеноксиацетонитрила (0,24 моля) и 37 г 2-нитронафталина (0,213 моля) в 200 мл диметилформамида. Через 2 ч при температуре -5 С выливали смесь в 4 л воды, содержащей 1 л концентрированной соляной кислоты, и экстрагировали водную фазу с помощью 3 х 500 мл дихлорметана. Промывали органическую фазу с помощью 300 мл воды, высушивали ее над сульфатом магния, фильтровали, а потом выпаривали растворитель. Получали 65 г продукта. Полученные 65 г продукта подвергали перекристаллизации из смеси циклогексан/этилацетат: 50/50% об./об. Этап В: 3H-бензо[е]индол При комнатной температуре и давлении водорода 4 бара подвергали гидрированию 33 г (2-нитро-1 нафтил)ацетонитрила (0,155 моля), растворенного в 630 мл этанола, содержащего 10% воды и 6,3 мл чистой уксусной кислоты, при использовании 19 г 10% палладия-на-углероде. После завершения процесса абсорбции отфильтровывали катализатор, концентрировали растворитель в вакууме, а потом переносили остаток в 250 мл дихлорметана, промывали органическую фазу с помощью 100 мл 0,1 N раствора гидроокиси калия, а потом высушивали органическую фазу над сульфатом магния, фильтровали и концентрировали. Остаток очищали с помощью хроматографии с использованием силикагеля, при этом в качестве элюанта использовали смесь циклогексан/этилацетат: 80/20: об./об. Этап С: 2,3-дигидро-1 Н-бензо[е]индол 10 г (0,06 моля) соединения, полученного на предыдущем этапе, растворяли в 50 мл тетрагидрофурана. К полученному раствору при температуре 0 С, прибавляли 120 мл комплекса боран/ТГФ в виде 1 М раствора в тетрагидрофуране, а потом прибавляли 120 мл трифторуксусной кислоты. Через 30 мин при температуре 0 С прибавляли 6 мл воды, перемешивали в течение 15 мин, а потом концентрировали смесь до осушения. Остаток переносили в 200 мл дихлорметана и промывали с помощью 200 мл 1N раствора гидроокиси натрия. Органическую фазу высушивали над сульфатом магния, фильтровали и концентрировали. Приготовление 2: 2,3-дигидро-1H-бензо[f]индол Протокол эксперимента для восстановления 1H-бензо[f]индола является таким же, что и для приготовления 1, этапа С. Синтез исходного соединения 1H-бензо[f]индола описан в литературе [Tetrahedron,49, 33., 7353 (1993); Heterocycles, 24, 7, 1845, (1986)]. Приготовление 3: 2,3-дигидро-7H-бензо-[e]-индол-6-карбонитрил Этап А: N-(5-циано-2-нафтил)ацетамид К 25 г N-(5,6,7,8-тетрагидронафт-2-ил)ацетамида, охлажденного до температуры 0 С, последовательно прибавляли 70 мл чистого триметисилилцианида, а потом 30 г дихлордицианохинона в 70 мл ди-3 007231 хлорметана. Через 3 ч при комнатной температуре вновь прибавляли раствор 60 г дихлордицианохинона в 140 мл дихлорметана. Смесь подогревали при температуре 20 С в течение 12 ч, а потом подогревали при температуре 60 С в течение 8 ч. После нейтрализации с помощью насыщенного раствора гидрокарбоната натрия отделяли органическую фазу и промывали с помощью воды. Полученный после концентрирования остаток очищали с помощью хроматографии через использовании силикагель и при использовании смеси циклогексан/этилацетат: 80:20: об./об. в качестве элюанта. Этап В: N-(1-бром-5-циано-2-нафтил)ацетамид К раствору 50 г (0,238 моля ) продукта, синтезированного на предыдущем этапе (при этом раствор был охлажден до температуры -5 С) в 500 мл дихлорметана и 25 мл пиридина, прибавляли 39,8 г брома,растворенного в 200 мл дихлорметана. Потом энергично перемешивали в течение 4 ч при комнатной температуре; последовательно разводили с помощью 500 мл дихлорметана, промывали органическую фазу дважды с помощью 300 мл воды, высушивали и концентрировали. Остаток перекристаллизовывали из смеси дихлорметан/метанол: 50/50: об./об. Этап С: 6-амино-5-бром-1-нафтонитрил Подогревали при температуре 80 С в течение 6 ч смесь 12,5 г (0,043 моля) продукта, синтезированного на предыдущем этапе, 3,6 г гидроокиси натрия, 195 мл метанола и 65 мл воды. После выпаривания метанола водную фазу экстрагировали дважды с помощью дихлорметана. Позднее последовательно высушивали и выпаривали. Остаток кристаллизовали из смеси дихлорметан/метанол. Этап D: Этил-1-бром-5-циано-2-нафтилкарбамат При температуре 0 С прибавляли 19 мл этилхлороформата к раствору 33 г (0,133 моля) продукта,синтезированного на предыдущем этапе, в 200 мл пиридина. Через 1 ч при температуре 5 С выпаривали растворитель, переносили остаток в 500 мл дихлорметана, три раза промывали органическую фазу с помощью 100 мл 0,1N соляной кислоты, а потом с помощью 200 мл 10% гидрокарбамата натрия, и в завершение - один раз водой. Остаток, полученный выпариванием, перекристаллизовывали из смеси дихлорметана/метанола. Этап Е: Этил-5-циано-1-[(триметилсилил)этинил-2-нафтилкарбамат В стальном реакторе перемешивали 14,1 г (0,044 моля) продукта, синтезированного на предыдущем этапе, 11 мл триметилсилилацетилена, 13 мл триэтиламина, 670 мг йодида меди и 1,54 г дихлорбис(трифенилфосфин)палладия. Потом закрывали реактор и подогревали реакционную смесь при температуре 80 С в течение 4 ч. Смесь разводили с помощью 200 мл дихлорметана и 100 мл воды, фильтровали смесь, отделяли органическую фазу, высушивали ее и выпаривали растворитель в вакууме. Полученный остаток очищали с помощью хроматографии при использовании силикагеля, применяя смесь циклогексан/этилацетат: 90/10: об./об. в качестве элюанта, после этого проводили кристаллизацию из того же растворителя. Этап F: 3H-бензо[е]индол-6-карбонитрил К раствору 2,74 г натрия в 280 мл сухого этанола прибавляли 10 г (0,0297 моля) продукта, синтезированного на предыдущем этапе, и подогревали смесь при температуре кипения с обратным холодильником в течение 1 ч. После выпаривания растворителя остаток переносили в 200 мл дихлорметана и органическую фазу промывали с помощью 200 мл воды. После выпаривания остаток очищали с помощью хроматографии через силикагель при использовании смеси циклогексан/этилацетат: 80/20: об./об. в качестве элюанта. Этап G: 2,3-дигидро-1 Н-бензо[е]индол-6-карбонитрил Пропись эксперимента для восстановления 3H-бензо[е]индол-6-карбонитрила была такой же, как и в приготовлении 1, этап С. Приготовление 4: 7-метокси-2,3-дигидро-1 Н-бензо[е]индол Этап А: 6-метокси-3,4-дигидро-1 (2 Н)нафталиноноксим Растворяли 100 г (0,57 моля) 6-метокси-1-тетралона в 2,5 л смеси этанол/вода: 80/20. Потом при комнатной температуре прибавляли 85 г (1,04 моля) ацетата натрия и 43 г (0,62 моля ) гидрохлорида гидроксиламина. Подогревали суспензию при температуре кипения с обратным холодильником в течение 4 ч. Разводили смесь с помощью 5 л воды и экстрагировали с помощью этилового эфира, промывали водой, высушивали над сульфатом магния и фильтровали. После выпаривания растворителя получали 99 г твердого вещества бежевого цвета. Этап В: 2-амино-6-метоси-5, 4-дигидро-1 (Н)-нафталинон Растворяли 50 г (0,26 моля) продукта, синтезированного на предыдущем этапе, в 185 мл пиридина,а потом прибавляли при комнатной температуре 54,9 г (0,29 моля) хлорида паратолуолсульфонила. Через 24 ч выливали смесь на лед, а потом отфильтровывали преципитат. Переносили преципитат в дихлорметан и промывали водой; высушивали органическую фазу над сульфатом магния и фильтровали. После выпаривания органического растворителя получали 89 г твердого вещества желтого цвета (промежуточный продукт 1). Прибавляли 7,48 г (0,32 моля) натрия к смеси толуол/этанол: 720/148 мл. После растворения разводили с помощью 940 мл толуола и быстро прибавляли 117 г (0,34 моля) промежуточного продукта 1. Че-4 007231 рез 24 ч при комнатной температуре отфильтровывали остаток паратолуолсульфоната натрия, промывали с помощью толуола, а потом выливали органический раствор в 10% раствор соляной кислоты (1,1 л). Отделяли продукт, один раз экстрагировали водой, а потом выпаривали водную фазу. Переносили остаток в этанол, а потом отфильтровывали преципитат. Получали 46,5 г твердого вещества бежевого цвета в форме гидрохлорида. Этап С: N-этил-N'-(6-метокси-1-оксо-1,2,3,4-тетрагидро-2-нафтил)мочевина Выливали 4,77 г (0,044 моля) этихлорформата при температуре 0 С в 5 г (0,022 моля) 2-амино-6 метокси-3, 4-дигидро-2H-нафталин-1-она, растворенного в пиридине. Через два часа при комнатной температуре концентрировали пиридин, переносили в дихлорметан, а потом промывали органическую фазу с помощью 0,1N раствора соляной кислоты, а потом с помощью насыщенного раствора гидрокарбоната натрия и воды; высушивали над сульфатом магния, фильтровали, и выпаривали растворитель. Получали 5,48 г твердого вещества оранжевого цвета. Этап D: N-этил-N'-(6-метокси-1,2,3,4-тетрагидро-2-нафтил)мочевина При температуре 60 С и в условиях атмосферного давления подвергали гидрированию 98 г (0,37 моля) продукта, полученного на этапе С, после чего растворяли его в 1,5 л этанола при использовании 10 г 5% палладия-на-углероде. После того, как абсорбция была завершена, отфильтровали катализатор и концентрировали растворитель в вакууме. Получали 88,2 г вещества в виде масла. Этап Е: N-этил-N'-(6-метокси-2-нафтил)мочевина Растворяли 7,42 г (0,0298 моля) продукта, синтезированного на предыдущем этапе, в 100 мл толуола. Прибавляли 13,51 г (0,0595 моля) дихлордицианохинона и подогревали при температуре кипения с обратным холодильником в течение 30 мин. Отфильтровывали преципитат при комнатной температуре,промывали с помощью толуола, а потом выпаривали растворитель. Остаток очищали с помощью хроматографии через силикагель при использовании чистого дихлорметана в качестве элюанта. При этом получали 4 г твердого вещества серого цвета. Этап F: 6-метокси-2-нафтиламин Растворяли 3,5 г продукта, синтезированного на предыдущем этапе, в 65 мл этанола, а потом прибавляли раствор гидроокиси натрия в 65 мл воды. После кипячения с обратным холодильником в течение 4 ч отфильтровывали при комнатной температуре образовавшийся преципитат. Переносили сырьевой продукт в дихлорметан и промывали водой до получения нейтральной реакции; высушивали над сульфатом магния, отфильтровывали преципитат, а потом выпаривали растворитель. Получали 2,02 г твердого вещества оранжевого цвета. Этап G: 1-йод-6-метокси-2-нафтиламин 28,2 г (0,16 моля) продукта, синтезированного на предыдущем этапе, растворяли в смеси дихлорметан/метанол: 1500/620 мл. К полученному раствору прибавляли при комнатной температуре 56,7 г (0,16 моля) дигидрохлориодата бензилтриметиламмония и 21,2 г (0,212 моля) карбоната кальция. Через 30 мин отфильтровывали нерастворенный материал, потом переносили органическую фазу в 10%-ный раствор бисульфита натрия и экстрагировали с помощью этилового эфира. Высушивали над сульфатом магния,фильтровали, а потом выпаривали. Остаток очищали с помощью хроматографии через силикагель при использовании смеси циклогексан/этиацетат: 70/30: об./об. в качестве элюанта. Этап Н: Этил 1-йод-6-метокси-2-нафтилкарбамат Превращение 1-йод-6-метокси-2-нафтиламина осуществляли при использовании способа, описанного в этапе С. Этап I: Этил 6-метокси-1-[(триметилсилил)этинил]-2-нафтилкарбамат Превращение этил-1-йод-6-метокси-2-нафтилкарбамата осуществляли при использовании способа,описанного в приготовлении 3, этап Е. Этап J: 7-метокси-3H-бензо[е]индол Превращение соединения, полученного на предыдущем этапе, осуществляли при использовании способа, описанного в приготовлении 3, этап F. Этап К: 7-метокси-2,3-дигидро-1 Н-бензо[е]индол Пропись для проведения восстановления 7-метокси-3H-бензо[е]индола является такой же, как и для приготовления 1, этап С. Приготовление 5: 4-метокси-3-(4-метил-1-пиперазинил)бензоилазид При комнатной температуре прибавляли раствор 5,3 г (0.025 моля) фенилдихлорфосфата в 100 мл дихлорметана к раствору 5 г (0.02 моля) 4-метокси-3-(4-метилпиперазин-1-ил)бензойной кислоты [J.Med. Chem.,32 (15), 2255 (1994)] и 3,25 г (0,05 моля) азида натрия в 4,05 мл пиридина. После перемешивания в течение 12 ч промывали органическую фазу с помощью 100 мл воды, отделяли органическую фазу, высушивали над сульфатом магния и выпаривали растворитель в вакууме при температуре 30 С. Пример 1. ГидрохлоридN-[4-метокси-3-(4-метил-1-пиперазинил)фенил]-1,2-дигидро-3Hбензо[е]индол-3-карбоксамида Подогревали при температуре кипения с обратным холодильником раствор 29 г (0,105 моля) соединения, синтезированного в приготовлении 5, в 400 мл толуола, потом охлаждали до температуры 20 С и прибавляли 100 мл дихлорметана, а потом прибавляли раствор 18 г (0,105 моля) продукта, полученного в-5 007231 приготовлении 1. После перемешивания в течение 24 ч при температуре 20 С выпаривали растворитель и очищали остаток с помощью хроматографии через силикагель при использовании смеси дихлорметан/метанол/аммоний: 95/5/0,5: об./об./об. в качестве элюанта. Основание потом превращали в соль с помощью соляной кислоты в растворе этанола. Точка плавления: 176-178 С. ГидрохлоридN-[4-метокси-3-(4-метил-1-пиперазинил)фенил]-2,3-дигидро-1HПример 2. бензо[е]индол-1-карбоксамида Пропись проведения эксперимента была такой же, как и для примера 1, при этом начинали с продукта приготовления 2. Пример 3. Гидрохлорид 6-циано-N-[4-метокси-3-(4-метил-1-пиперазинил)-фенил]-1,2-дигидро-3Hбензо[е]-индол-3-карбоксамида Пропись проведения эксперимента была такой же, как и для примера 1, при этом начинали с продукта приготовления 3. Пример 4. Гидрохлорид 7-метокси-N-[4-метокси-3-(4-метил-1-пиперазинил)фенил]-1,2-дигидро-3Hбензо[е]-индол-3-карбоксамида Пропись проведения эксперимента была такой же, как и для примера 1, при этом начинали с продукта приготовления 4. Фармакологические исследования Пример А: Определение аффинности для 2-адренергических рецепторов у крыс Аффинность определяли при проведении конкурентных экспериментов с [3H]-RX 821,002. Мембраны готовили из церебральной коры головного мозга крыс и инкубировали в трехкратной повторности с 0,4 нМ [3H]-RX 821,002 и исследуемым соединением в заключительном объеме 1,0 мл в течение 60 мин при температуре 22 С. Буфер для инкубации содержал 50 нМ Трис-HCl (рН 7,5), 1 мМ ЕДТА и 100 мкМGppNHp. Неспецифическое связывание определяли при использовании 10 мкМ фентоламина. Анализ данных: В конце инкубации инкубационную среду фильтровали через фильтры WHATMANGF/B, импрегнированные 0,1% полиэтиламином, и промывали трижды с помощью 5 мл охлажденного буфера. Оставшуюся на фильтрах радиоактивность определяли с помощью подсчета жидкостной сцинтиляции. Изотермы связывания анализировали при использовании нелинейной регрессии. Результаты: Соединения в соответствии с изобретением демонстрировали специфическую антагонистическую активность в отношении 2-адренергического рецептора, например, для соединения примера 1 pKi равно 7,4, в то время, как для сравнительного продукта (пример 11 заявки ЕР-1170288), 6-хлор-5 фтор-N-[4-метокси-3-(4-метил-1-пиперазинил)фенил]-1-индолинкарбоксамид, значение pKi составляло 6,4. Пример В: Определение аффинности для 5-НТ 2 С Аффинность соединений в соответствии с изобретением определяли путем проведения конкурентных экспериментов в присутствии [Н]-мезулергина в буфере для инкубации, содержащем Hepes 20 мМ,EDTA 2 мМ, 0,1% аскорбиновой кислоты (рН = 7,7), при температуре 22 С. Константа диссоциацииKD[Н]-мезулергина составляла 0,54 мМ. Неспецифическую фракцию определяли в присутствии 1 мкМ миансерина, последний является стандартным продуктом для каждого эксперимента. В конце инкубационного периода раствор фильтровали через фильтры GF/B-Unifilter, предварительно обработанные с помощью PEI (0,1%), после этого трижды промывали с помощью буфера для инкубации. Оставшуюся на фильтрах радиоактивность определяли с помощью подсчета жидкостной сцинтиляции. Изотермы связывания анализировали с помощью нелинейной регрессии для определения значений IC50. Их превращали в значения рКi (где Ki представляет собой константу диссоциации). Было выявлено, что соединения в соответствии с изобретением имеют высокую аффинность для рецепторов 5-НТ 2C; например, соединение в соответствии с примером имело значение рКi, 8,2, в то время, как сравнительное соединение (пример 11 заявки ЕР-1170288), 6-хлор-5-фтор-N-[4-метокси-3-(4 метил-1-пиперазинил)фенил]-1-индолинкарбоксамид, имел значение рКi 7,4. Пример С: Измерение нейромедиаторов в лобной коре головного мозга крыс Диализ Осуществляли хирургию с помощью анестезии, индуцированной пентобарбиталом (60 мг/кг, интраперитонеально). Самцов крыс Wistar весом 200 - 220 г (Iffa Credo, Arbresle, France) помещали стереотаксическое устройство и вводили проводниковую канюлю (CMAMicrodialyse AB, Stockholm, Sweden) в лобную кору при координатах (в мм): передне-заднее: + 2,2, латеральное: + 0,6, вентральное отклонение:- 0,2. Крыс помещали в отдельные клетки и восстанавливали после анестезии в течение 5 дней. В день проведения диализа вводили зонд Cuprophan СМА/11 (длина: 4 мм; внешний диаметр: 0,24 мм) в проводниковую канюлю, после чего проводили перфузию при скорости 1 мкл/мин раствора следующего состава: NaCl: 147,2 мМ; КСl: 4 мМ; СаСl2: 2,3 мМ, значение рН доводили до 7,3 с помощью фосфатного буфера. Через два часа после имплантации образцы диализата собирали каждые 20 мин в течение 4 ч. Собирали три основных образца перед введением лекарственного средства.-6 007231 Хроматография Норадреналин (NA) и допамин (DA) измеряли так, как описано ниже: 20 мкл диализата образца разводили с помощью 20 мкл мобильной фазы (NaH2PO4: 75 мМ, ЭДТА: 20 мкМ, декансульфонат натрия: 1 мМ, метанол: 17,5%, триэтиламин: 0,01%, рН: 5,70) и 33 мкл подвергали анализу с помощью ВЭЖХ при использовании для разделения колонки с обратной фазой (Hypersil С 18, 150 х 4,6 мм; размер частиц 5 мкм) с термостатическим контролем при температуре 43 С, для количественной оценки использовали кулонометрический детектор (ESA5014, Coulochem II, ESA, Chelmsford, USA). Потенциал первого электрода составлял -90 мВ (восстановление), а таковой второго + 280 мВ (окисление). Скорость истечения мобильной фазы составляла 2 мл/мин. Предел чувствительности для NA и DA составлял 0,2 пг. Ацетонитрил (ACh) измеряли и количественно оценивали при отсутствии ингибитора ацетонитрилэстеразы (AChE). Аликвоты 20 мкл собирали в 10 мкл 0,01% уксусной кислоты. Аликвоты объемом 20 мкл подвергали анализу с помощью ВЭЖХ. Мобильная фаза состояла из Na2HPO4: 50 мМ и ProClin: 0,5% (BAS, Congleton, UK), pH 8,2. Стационарную фазу компоновали из катионообменной колонки(Sepstik, 530 х 1,0 мм, размер частиц 10 мкм, ВAS), предколонки (ферментативный реактор холиноксидаза/каталаза, 55 х 1 мм, BAS) и последующей колонки (ферментативный реактор холиноксидаза/AChE, 50 х 1 мм, BAS). Систему поддерживали при температуре 35 С. Количественную оценку осуществляли с помощью амперометрического детектора (LC-4B, BAS). На стеклянном углеродном рабочем электроде(MF2098, BAS) размещался слой пероксидаза-редокс полимера. Потенциал этого электрода составлял+100 мВ относительно сравнительного электрода Ag/AgCl. Скорость истечения мобильной фазы составляла 0,14 мл/мин. Предел чувствительности для ACh составлял 0,1 пг. В качестве примера, соединение примера 1 (10,0 мг/кг, подкожно) вызывало существенное увеличение внеклеточных концентраций NA, DA и ACh в диализатах, взятых из лобной коры головного мозга крыс, находящихся в сознании (таблица 1). Сравниваемое соединение (пример 11 заявки ЕР-1170288), 6 хлор-5-фтор-N- [4-метокси-3-(4-метил-1-пиперазинил)фенил]-1-индолинкарбоксамид (10,0 мг/кг, подкожно), вызывал весьма умеренное увеличение внеклеточных концентраций NA и DA и не вызывал изменения внеклеточной концентрации ACh (таблица 1). Уровни NA, DA и ACh выражали как процент площади под кривой (% AbC)S.E.M. в моменты времени от 20 до 120 мин после введения лекарственного средства. Сравнивали эффекты соединения примера 1 (ANOVA) с теми, что были получены на обработанных с помощью растворителя животных или на животных обработанных с помощью 6-хлор-5 фтор-N-[4-метокси-3-(4-метил-1-пиперазинил)фенил]-1-индолинкарбоксамида. Таблица 1:Р 0,005 против растворителя и b: Р 0,05 против 6-хлор-5-фтор-N-[4-метокси-3-(4-метилпиперазинил)фенил]-1-индолинкарбоксамид. Пример D: Фармацевтическая композиция Формула для приготовления 1000 таблеток, каждая из которых содержит 10 мг активного ингредиента Соединения в соответствии с примером 1 10 г Гидроксипропилцеллюлоза 2 г Пшеничный крахмал 10 г Лактоза 100 г Стеарат магния 3 г Тальк 3 г в которой R1 и R2 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино, диалкиламино или группой трифторметила, a R3 и R4 представляют собой атом водорода, илиR1 и R4 представляют собой атом водорода, a R2 и R3 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино,диалкиламино или группой трифторметила, илиR1 и R2 представляют собой атом водорода, а R3 и R4 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино,диалкиламино или группой трифторметила,их энантиомеры и диастереоизомеры, их аддитивные соли с фармацевтически приемлемой кислотой или основанием,при этом понятно, что термин "алкил" означает неразветвленную или разветвленную углеводородную цепь, содержащую от 1 до 6 атомов углерода,термин "алкокси" означает неразветвленную или разветвленную группу алкилокси, содержащую от 1 до 6 атомов углерода. 2. Соединения формулы (I) в соответствии с п.1, в которых R1 и R2 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино, диалкиламино или группой трифторметила, a R3 и R4 представляют собой атом водорода, их энантиомеры и диастереоизомеры, а также их аддитивные соли с фармацевтически приемлемой кислотой или основанием. 3. Соединение формулы (I) в соответствии с п.1 или 2, которое представляет собой N-[4-метокси-3(4-метил-1-пиперазинил)фенил]-1,2-дигидро-3H-бензо[е]индол-3-карбоксамид, а также его аддитивные соли с фармацевтически приемлемой кислотой или основанием. 4. Способ получения соединения формулы (I) в соответствии с п.1, отличающийся тем, что в качестве исходного соединения используют бензоиндольное соединение формулы (II) в которой R1, R2, R3 и R4 являются такими, как определено для формулы (I), которое конденсируют с соединением формулы (III)(I), в которой Y представляют собой группу -N=C=O или -C(O)-N3, для получения соединения формулы которое может быть очищено, в случае необходимости, в соответствии с традиционным способом очистки,которое разделяют на изомеры (диастереоизомеры и энантиомеры), в случае необходимости, в соответствии с традиционным способом разделения,которое превращают, если это является желательным, в его аддитивные соли при использовании приемлемой кислоты или основания.-8 007231 5. Фармацевтические композиции, включающие в качестве активного ингредиента по крайней мере одно соединение в соответствии с любым из пп.1-3, отдельно или в комбинации с одним или более инертными, нетоксическими, фармацевтически приемлемыми наполнителями или носителями. 6. Фармацевтические композиции в соответствии с п.5, включающие по крайней мере один активный ингредиент в соответствии с любым из пп.1-3, для применения в качестве лекарственного средства с двойственной 2-AR/5-НТ 2C антагонистической активностью. 7. Фармацевтические композиции в соответствии с п.5, включающие по крайней мере один активный ингредиент в соответствии с любым из пп.1-3, для применения при производстве лекарственных средств для лечения депрессии, беспокойства, расстройств поведения, связанных с повышенной возбудимостью, шизофрении, болезни Паркинсона, когнитивных расстройств, расстройств либидо и сексуальных дисфункций, а также расстройств сна.
МПК / Метки
МПК: C07D 209/00, C07D 403/12, A61K 31/496, A61P 25/18
Метки: содержат, фармацевтические, новые, получения, бензоиндолина, соединения, которые, композиции, способ
Код ссылки
<a href="https://eas.patents.su/10-7231-novye-soedineniya-benzoindolina-sposob-ih-polucheniya-i-farmacevticheskie-kompozicii-kotorye-ih-soderzhat.html" rel="bookmark" title="База патентов Евразийского Союза">Новые соединения бензоиндолина, способ их получения и фармацевтические композиции, которые их содержат</a>
Соединения бензотиазина и бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 7227
Опубликовано: 25.08.2006
Авторы: Десо Патрис, Корди Алекс, Лестаж Пьер
МПК: A61K 31/549, A61P 25/00, A61K 31/5415...
Метки: которые, соединения, получения, содержат, бензотиазина, бензотиадиазина, композиции, способ, фармацевтические
Формула / Реферат:
1. Соединения формулы (I) где R1 представляет собой арильную или гетероарильную группу, R2 представляет собой атом водорода, атом галогена или гидроксильную группу, А представляет собой CR4R5 группу или NR4 группу, R3 представляет собой атом водорода, линейную или разветвленную С1-С6-алкильную группу или С3-С7-циклоалкильную группу, R4 представляет собой атом водорода или линейную или разветвленную С1-С6-алкильную группу, или А представляет...
Производные гидроксиалкилиндолкарбазола, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 6201
Опубликовано: 27.10.2005
Авторы: Бизо-Эспьяр Жан-Ги, Ренар Пьер, Пьер Ален, Хикман Джон, Пфайфер Брюно, Прюдом Мишель, Моро Паскаль, Марминон Кристель
МПК: A61P 25/00, C07H 19/23, A61K 31/7056...
Метки: способ, производные, содержат, получения, фармацевтические, композиции, которые, гидроксиалкилиндолкарбазола
Формула / Реферат:
1. Соединения формулы (I) в которой R1 и R2, которые могут быть одинаковыми или разными, каждый, независимо друг от друга, представляет собой группу, выбранную из водорода, линейного или разветвленного (C1-C6)алкила, арил(C1-C6)алкила, в котором алкильная часть может быть линейной или разветвленной, гидрокси, линейного или разветвленного (C1-C6)гидроксиалкила, линейного или разветвленного дигидрокси(C1-C6)алкила, линейного или разветвленного...
Новые соединения имидазолина, способ их получения и содержащие их фармацевтические композиции

Номер патента: 4536
Опубликовано: 24.06.2004
Авторы: Корди Алекс, Ньюмен-Танкреди Адриан, Миллан Марк, Гобер Алан, Лякост Жан-Мишель
МПК: C07D 233/06, A61P 25/24, A61K 31/4164...
Метки: содержащие, способ, соединения, получения, новые, композиции, фармацевтические, имидазолина
Формула / Реферат:
1. Соединения формулы (I) где A представляет кольцо бензола, незамещенное или замещенное 1-4 одинаковыми или различными группами, выбранными из неразветвленного или разветвленного (C1-C6)алкила, неразветвленного или разветвленного (C1-C6)алкокси, гидрокси, полигалоген (C1-C6)алкила, у которого алкильная часть не разветвлена или разветвлена, циано, нитро, амино, алкиламино, диалкиламино, тиоалкила, сульфонилалкила, сульфинилалкила, карбокси,...
Новые соединения бензо [b]пирано[3,2-h]акридин-7-она, способ их получения и содержащие их фармацевтические композиции

Номер патента: 5374
Опубликовано: 24.02.2005
Авторы: Ренар Пьер, Сеген Элизабет, Пьер Ален, Хикмен Джон, Кош Мишель, Пфейффер Брюно, Эль Омри Абдельхаким, Тийекен Франсуа, Мишель Сильви
МПК: A61P 35/00, C07D 221/04, A61K 31/4741...
Метки: фармацевтические, получения, новые, способ, композиции, содержащие, b]пирано[3,2-h]акридин-7-она, бензо, соединения
Формула / Реферат:
1. Соединения формулы (I) в которой X и Y, которые могут быть одинаковыми или различными, представляют собой независимо друг от друга группу, выбранную из атомов водорода или галогена, меркапто, циано, нитро, линейной или разветвленной (C1-C6)алкильной, линейной или разветвленной (C1-C6)тригалоалкильной и линейной или разветвленной тригало-(C1-C6)алкилкарбониламиногруппы, групп формул -ORa, -NRaRb, -NRa-C(O)-T1, -O-C(O)-T1, -OT2-NRaRb,...
Новые циклические соединения, имеющие циклоалкиленовую цепь, способ их получения и включающие их фармацевтические композиции

Номер патента: 2818
Опубликовано: 31.10.2002
Авторы: Мат-Алленма Моник, Беннежан Каролин, Лефа-Ле Галь Мари, Ланглуа Мишель, Ренар Пьер, Делагранж Филипп
МПК: A61K 31/343, C07D 307/80, C07C 233/06...
Метки: включающие, способ, цепь, циклоалкиленовую, композиции, получения, циклические, имеющие, фармацевтические, соединения, новые
Формула / Реферат:
1. Соединение формулы (I) где А представляет собой - кольцевую систему формулы (II) где R может быть заместителем в любом положении кольцевой системы II, - или А представляет кольцевую систему формулы (III) где Z представляет собой атом кислорода, атом серы или группу NR1 (где R1 представляет собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу, D' представляет бензольное кольцо или пиридиновое кольцо, где R замещает D'...
Предыдущий патент: Способ получения скопиновых эфиров
Следующий патент: Иммуностимулирующие олигонуклеотиды, содержащие комбинацию мотивов и обладающие повышенной активностью
Случайный патент: Способ получения чистого кремния