Перекрестно-сшитые антибиотики “гликопептид-цефалоспорин”
Номер патента: 7001
Опубликовано: 30.06.2006
Авторы: Фазери Пол, Тернер С.Дерек, Маркесс Дэниэл, Моран Эдмунд Дж., Эгжен Джеймс, Линселл Мартин С., Нодуэлл Мэттью Б., Лонг Дэниэл Д.




























Формула / Реферат
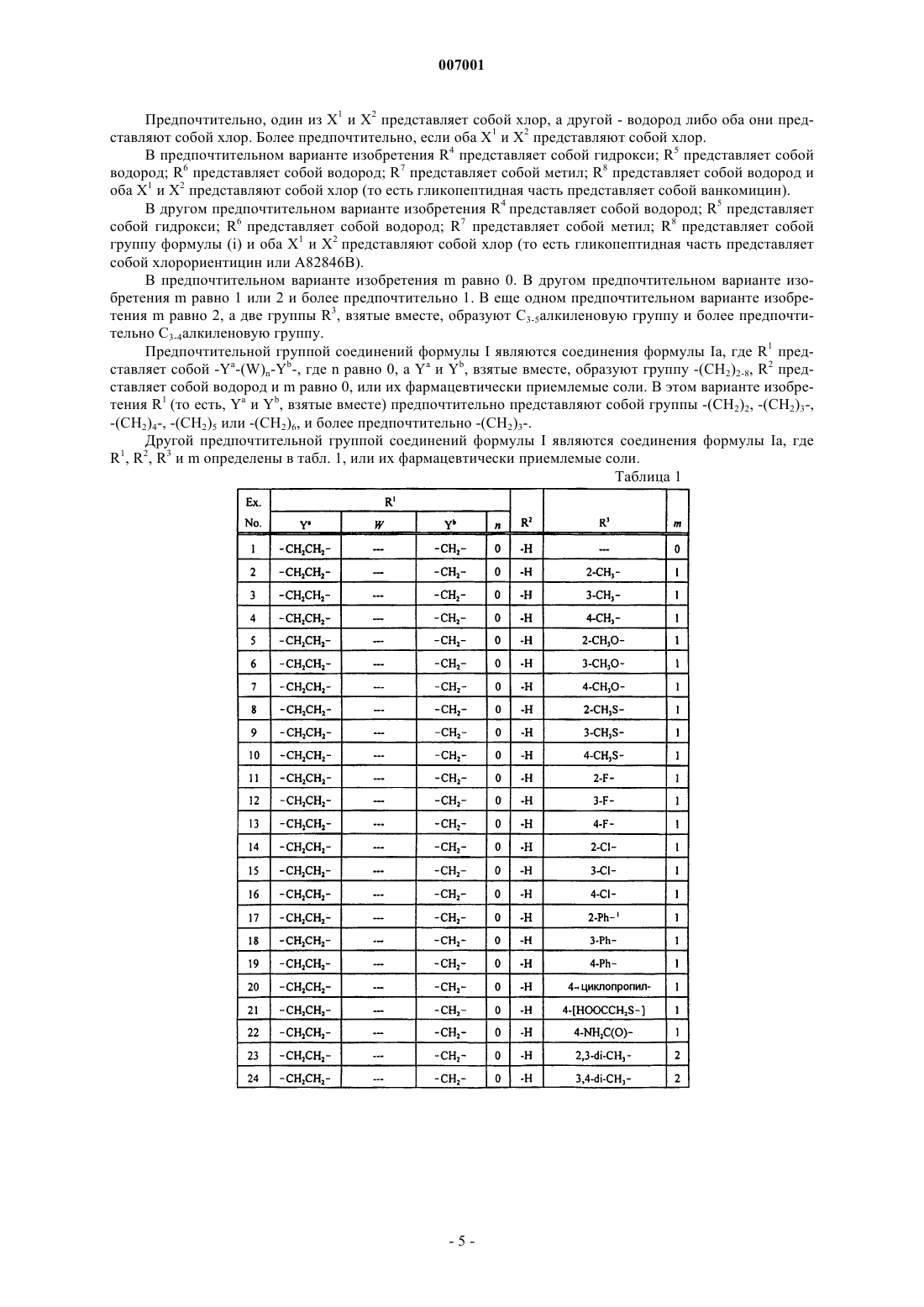
1. Соединение формулы I

или его фармацевтически приемлемая соль,
где X1 и X2 независимо выбраны из группы, состоящей из водорода и хлора;
R1 представляет собой -Ya-(W)n-Yb-;
W представляет собой -О- или С6-10арилен;
Ya и Yb независимо представляют собой C1-6алкилен;
R2, R5, R6 и R8 представляют собой водород;
каждый R3 независимо выбран из группы, состоящей из C1-6алкила, С2-6алкенила, С2-6алкинила, С3-6 циклоалкила, С6-10арила, С2-9гетероарила, С3-6гетероциклила и Ra, либо две смежные группы R3, взятые вместе, образуют С3-6алкилен или -О-(C1-6алкилен)-О-, где каждая алкильная, алкиленовая, алкенильная и алкинильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Ra и Rc; и каждая арильная, циклоалкильная, гетероарильная и гетероциклическая группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Rb;
где каждый Ra независимо выбран из группы, состоящей из -ORd, галогена, -SRd, -S(O)Rd, -S(O)2Rd, -S(О)2ORd, -S(О)2NRdRe, -NRdRe, -CO2Rd, -ОС(O)Rd, -C(O)NRdRe, -NRdC(O)Re, -OC(O)NRdRe, -NRdC(O)ORe, -NRdC(O)NRdRe, -СF3 и -ОСF3;
каждый Rb независимо выбран из группы, состоящей из C1-6алкила, С2-6алкенила, С2-6алкинила и Ra;
каждый Rc независимо выбран из группы, состоящей из С3-6циклоалкила, С6-10арила, С2-9гетероарила и С3-6гетероциклила, где каждая циклоалкильная, арильная, гетероарильная и гетероциклильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из C1-6алкила и Rf;
каждый Rd и Re независимо выбран из группы, состоящей из водорода, C1-6алкила, С2-6алкенила, С2-6 алкинила, С3-6циклоалкила, С6-10арила, С12-9гетероарила и С3-6гетероциклила, либо Rd и Re, взятые вместе с атомами, с которыми они связаны, образуют С3-6гетероциклическое кольцо, имеющее 1-3 гетероатома, независимо выбранных из кислорода, азота или серы, где каждая алкильная, алкенильная и алкинильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Rc и Rf, и каждая арильная, циклоалкильная, гетероарильная и гетероциклильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из C1-6алкила и Rf;
каждый Rf независимо выбран из группы, состоящей из -ОН, -OC1-6алкила, -SC1-6алкила, -F, -Cl, -NH2, -NН(С1-6алкила), -N(C1-6алкила)2, -ОС(О)C1-6алкила, -С(O)ОС1-6алкила, -NHC(О)C1-6алкила, -С(О)ОН, -С(О)NH2, -С(O)NHC1-6алкила, -С(О)N(C1-6алкила)2, -СF3 и -ОСF3;
где указанные гетероарильная и гетероциклильная группы содержат от 1 до 3 гетероатомов, выбранных из азота, кислорода или серы;
R4 представляет собой гидрокси;
R7 представляет собой метил;
m равно 0, 1 или 2 и
n равно 0 или 1.
2. Соединение по п.1, где n равно 0.
3. Соединение по п.1, где n равно 0 и Ya и Yb, взятые вместе, образуют группу -(СН2)2-8-.
4. Соединение по п.3, где n равно 0 и Ya и Yb, взятые вместе, образуют группы -(СН2)2-, -(СН2)3-, -(СН2)4-, -(СН2)5- или -(СН2)6-.
5. Соединение по п.4, где n равно 0 и Ya и Yb, взятые вместе, образуют группу -(СН2)3-.
6. Соединение по п.1, где n равно 1.
7. Соединение по п.1, где n равно 1 и Ya и Yb оба представляют собой -СН2-.
8. Соединение по п.7, где W представляет собой фенилен.
9. Соединение по п.1, где n равно 1, Ya и Yb оба представляют собой -СН2СН2- и W представляет собой -О-.
10. Соединение по любому из пп.1-9, где m равно 0.
11. Соединение по любому из пп.1-9, где m равно 1 или 2 и каждый R3 независимо выбран из группы, состоящей из C1-6алкила, С3-6циклоалкила, -ORd, -SRd, -F или -Сl; либо две смежные группы R3, взятые вместе, образуют С3-6алкилен.
12. Соединение по любому из пп.1-11, где оба X1 и X2 представляют собой хлор.
13. Соединение по п.1, где R1 представляет собой -Ya-(W)n-Yb-, где n равно 0 и Ya и Yb, взятые вместе, образуют группу -(СН2)3-, оба X1 и X2 представляют собой хлор и m равно 0.
14. Соединение по п.1, где оба X1 и X2 представляют собой хлор и R1, R2, R3 и m такие, как определено в табл. 1.
15. Соединение формулы II

или его соль,
где Р1 и Р2 независимо представляют собой водород или аминозащитную группу;
Р3 представляет собой водород или карбоксизащитную группу;
Q представляет собой удаляемую группу или группу формулы

где R1 представляет собой -Ya-(W)n-Yb-;
W представляет собой -О- или С6-10арилен;
Ya и Yb независимо представляют собой C1-6алкилен;
R2 представляет собой водород;
каждый R3 независимо выбран из группы, состоящей из C1-6алкила, С2-6алкенила, С2-6алкинила, С3-6 циклоалкила, С6-10арила, С2-9гетероарила, С3-6гетероциклила и Ra, либо две смежные группы R3, взятые вместе, образуют С3-6алкилен или -О-(C1-6алкилен)-О-, где каждая алкильная, алкиленовая, алкенильная и алкинильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Ra и Rc; и каждая арильная, циклоалкильная, гетероарильная и гетероциклическая группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Rb;
каждый Ra независимо выбран из группы, состоящей из -ORd, галогена, -SRd, -S(O)Rd, -S(O)2Rd, -S(O)2ORd, -S(O)2NRdRe, -NRdRe, -CO2Rd, -OC(O)Rd, -C(O)NRdRe, -NRdC(O)Re, -OC(O)NRdRe, -NRdC(O)ORe, -NRdC(O)NRdRe, СF3 и -ОСF3;
каждый Rb независимо выбран из группы, состоящей из C1-6алкила, С2-6алкенила, С2-6алкинила и Ra;
кажфыщ Rc независимо выбран из группы, состоящей из С3-6циклоалкила, С6-10арила, С2-9гетероарила и С3-6гетероциклила, где каждая циклоалкильная, арильная, гетероарильная и гетероциклильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из C1-6 алкила и Rf;
каждый Rd и Re независимо выбран из группы, состоящей из водорода, C1-6алкила, С2-6алкенила, С2-6 алкинила, С3-6циклоалкила, С6-10арила, С2-9гетероарила и С3-6гетероциклила, либо Rd и Re, взятые вместе с атомами, с которыми они связаны, образуют С3-6гетероциклическое кольцо, имеющее 1-3 гетероатома, независимо выбранных из кислорода, азота или серы, где каждая алкильная, алкенильная и алкинильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Rc и Rf, и каждая арильная, циклоалкильная, гетероарильная и гетероциклильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из C1-6алкила и Rf;
каждый Rf независимо выбран из группы, состоящей из -ОН, -OC16алкила, -SC1-6алкила, -F, -Cl, -NH2, -NH(C1-6алкила), -N(С1-6алкила)2, -ОС(О)C1-6алкила, -С(О)OC1-6алкила, -NHC(O)С1-6алкила, -С(О)ОН, -C(O)NH2, -С(О)NHC1-6алкила, -С(О)N(C1-6алкила)2, -СF3 и -ОСF3;
Х- представляет собой необязательно присутствующий анион.
m равно 0, 1 или 2 и
n равно 0 или 1.
16. Соединение формулы III

или его соли,
где Р1 и Р2 независимо представляют собой водород или аминозащитную группу;
Р4 представляет собой водород или карбоксизащитную группу;
R1 представляет собой -Ya-(W)n-Yb-;
W представляет собой -О- или С6-10арилен;
Ya и Yb независимо представляют собой C1-6алкилен;
R2 представляет собой водород и
n равно 0 или 1.
17. Фармацевтическая композиция для лечения бактериальной инфекции у млекопитающего, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по любому из пп.1-14.
18. Способ лечения бактериальной инфекции у млекопитающего, где указанный способ предусматривает введение указанному млекопитающему фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по любому из пп.1-14.
19. Способ ингибирования роста бактерий, где указанный способ предусматривает контактирование бактерий с ингибирующим рост количеством соединения по любому из пп.1-14.
20. Способ ингибирования биосинтеза клеточной стенки бактерий, где указанный способ предусматривает контактирование бактерий с определенным количеством соединения по любому из пп.1-14, ингибирующим биосинтез клеточной стенки.
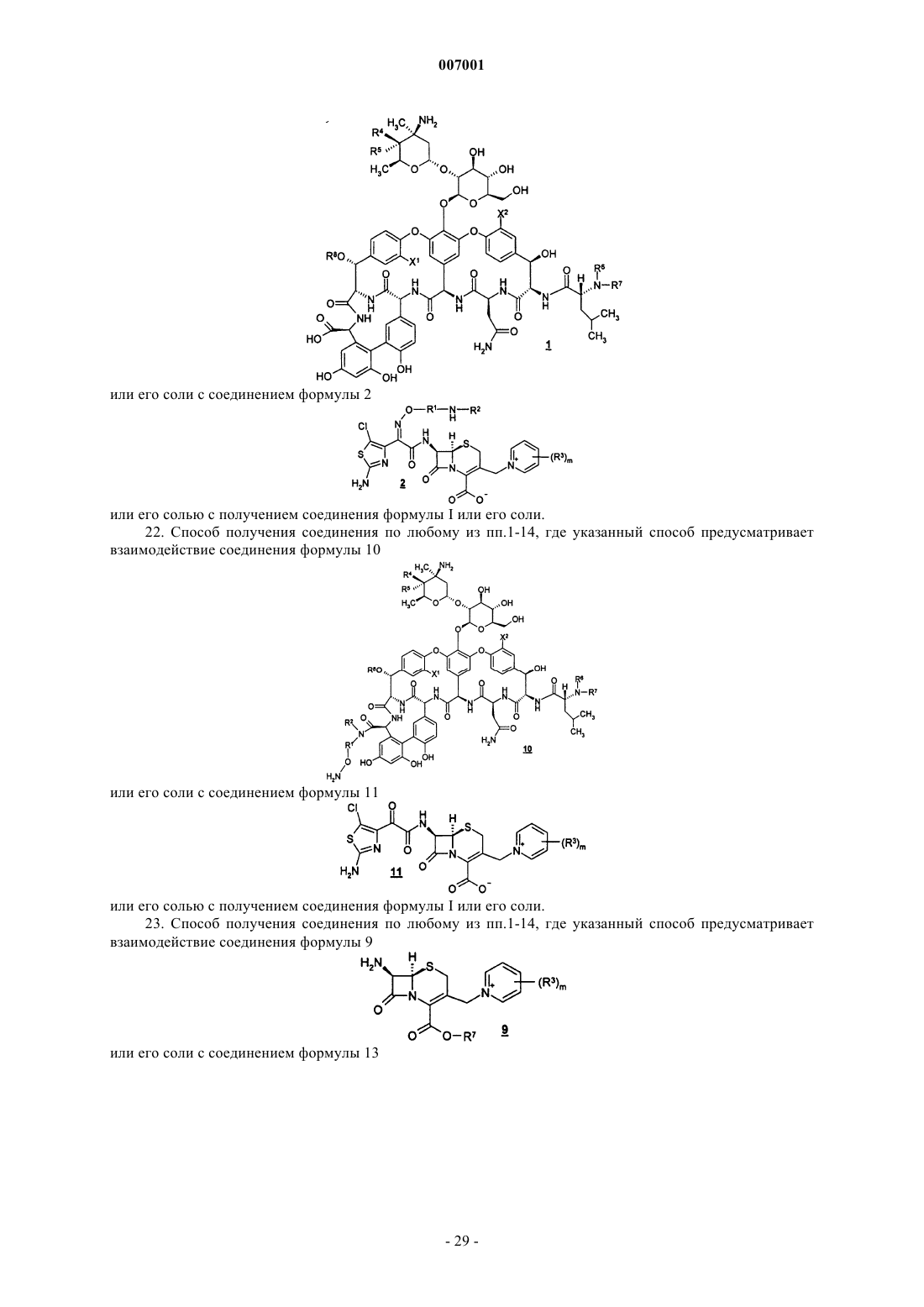
21. Способ получения соединения по любому из пп.1-14, где указанный способ предусматривает взаимодействие гликопептида формулы 1

или его соли с соединением формулы 2

или его солью с получением соединения формулы I или его соли.
22. Способ получения соединения по любому из пп.1-14, где указанный способ предусматривает взаимодействие соединения формулы 10

или его соли с соединением формулы 11

или его солью с получением соединения формулы I или его соли.
23. Способ получения соединения по любому из пп.1-14, где указанный способ предусматривает взаимодействие соединения формулы 9

или его соли с соединением формулы 13

или его солью с получением соединения формулы I или его соли.
24. Продукт, полученный способом по любому из пп.21-23.
25. Применение соединения по любому из пп.1-14, при приготовлении лекарственного средства для лечения бактериальной инфекции у млекопитающего.
Текст
007001 Предпосылки создания изобретения Область, к которой относится изобретение Настоящее изобретение относится к новым перекрестно-сшитым соединениям ванкомицинацефалоспорина, которые могут быть использованы в качестве антибиотиков. Настоящее изобретение также относится к фармацевтическим композициям, содержащим указанные соединения; к способам использования таких соединений в качестве антибактериальных агентов и к способам и промежуточным соединениям для получения указанных соединений. Уровень техники Специалистам известны различные классы соединений-антибиотиков, например -лактамовые антибиотики, такие как цефалоспорины, и гликопептидные антибиотики, такие как ванкомицин. Перекрестно-сшитые антибиотики также известны специалистам. См., например, патент США 5693791, выданный W.L. Truett и озаглавленный "Antibiotics and Process for Preparation", и WO 99/64049 A1, опубликованную 16 декабря 1999 и озаглавленную "Novel Antibacterial Agents". Несмотря на существование указанных соединений, необходимость в получении новых антибиотиков, обладающих улучшенными свойствами, например более высокой эффективностью против грамположительных бактерий, остается актуальной. В частности, необходимо получить новые антибиотики,которые обладали бы высокой эффективностью против штаммов бактерий, являющихся резистентными к антибиотикам, таких как резистентные к метициллину бактерии Staphylococci aureus (MRSA) и резистентные к метициллину бактерии Staphylococci epidermitis (MRSE). Краткое описание изобретения Настоящее изобретение относится к новым перекрестно-сшитым соединениям гликопептидацефалоспорина, которые могут быть использованы в качестве антибиотиков. Было неожиданно обнаружено, что помимо других свойств, соединения по настоящему изобретению обладают не предполагаемой ранее эффективностью против грамположительных бактерий, включая резистентные к метициллинуStaphylococci aureus (MRSA) и резистентные к метициллину Staphylococci epidermitis (MRSE). В соответствии с этим в одном из своих аспектов по композиции настоящее изобретение относится к соединению формулы I или к его фармацевтически приемлемой соли,где X1 и X2 независимо выбраны из группы, состоящей из водорода и хлора;W выбран из группы, состоящей из -О-, или С 6-10 арилена и где каждая ариленовая, циклоалкиленовая и гетероариленовая группа необязательно замещена 1-3 заместителями, независимо выбранными изYa и Yb независимо представляют собой C1-6 алкилен;R8, R5, R6, R2 представляют собой водород или C1-6 алкил; каждый R3 независимо выбран из группы, состоящей из C1-6 алкила, С 2-6 алкенила, С 2-6 алкинила, С 3-6 циклоалкила, С 6-10 арила, С 2-9 гетероарила, С 3-6 гетероциклила и Ra, либо две смежные группы R3, взятые вместе, образуют С 3-6 алкилен или -О-(C1-6 алкилен)-О-, где каждая алкильная, алкиленовая, алкенильная и алкинильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы,-1 007001 состоящей из Ra и Rc; и каждая арильная, циклоалкильная, гетероарильная и гетероциклическая группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Rb; каждый Ra независимо выбран из группы, состоящей из -ORd, галогена, -SRd, -S(O)Rd, -S(O)2Rd,-S(O)2ORd, -S(О)2NRdRe, -NRdRe-, -СO2Rd, -OC(O)Rd, -C(O)NRdRe, -NRdC(O)Re, -ОС(O)NRdRe,-NRdC(O)ORe, -NRdC(O)NRdRe, -СF3 и -ОСF3; каждый Rb независимо выбран из группы, состоящей из C1-6 алкила, С 2-6 алкенила, С 2-6 алкинила и Ra; каждый Rc независимо выбран из группы, состоящей из С 3-6 циклоалкила, С 6-10 арила, С 2-9 гетероарила и С 3-6 гетероциклила, где каждая из циклоалкильных, арильных, гетероарильных и гетероциклильных групп необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из C1-6 алкила и Rf; каждый Rd и Re независимо выбран из группы, состоящей из водорода, C1-6 алкила, С 2-6 алкенила, С 2-6 алкинила, С 3-6 циклоалкила, С 6-10 арила, С 2-9 гетероарила и С 3-6 гетероциклила, либо Rd и Re, взятые вместе с атомами, с которыми они связаны, образуют С 3-6-гетероциклическое кольцо, имеющее 1-3 гетероатома,независимо выбранных из кислорода, азота или серы, где каждая алкильная, алкенильная и алкинильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Rc и Rf, и каждая арильная, циклоалкильная, гетероарильная и гетероциклильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из C1-6 алкила и Rf; каждый Rf независимо выбран из группы, состоящей из -ОН, -OC1-6 алкила, -SC1-6 алкила, -F, -Cl,-NH2, -NH(C1-6 алкила), -N(С 1-6 алкила)2, -ОС(О)С 1-6 алкила, -С(О)OC1-6 алкила, NHC(O)C1-6 алкила,-С(О)ОН, -C(O)NH2, -С(О)NHC1-6 алкила, -С(О)N(C1-6 алкила)2, -СF3 и -ОСF3;R4 представляет собой гидрокси; R7 представляет собой метил; и где упомянутая гетероарильная или гетероциклильная группы включают в себя от 1 до 3 гетероатомов, выбранных из N, О и S.n равно 0 или 1. Настоящее изобретение также относится к промежуточным соединениям, используемым для получения соединений формулы I и их солей. В соответствии с этим, в одном из своих аспектов по композиции настоящее изобретение относится к соединению формулы II или к его соли,где R1 и R2 определены выше; Р 1 и Р 2 независимо представляют собой водород или аминозащитную группу; Р 3 представляет собой водород или карбоксизащитную группу;Q представляет собой удаляемую группу или группу формулы где R3 и m определены выше, и X- необязательно представляет собой анион; причем, указанные соединения могут быть использованы в качестве промежуточных соединений для получения соединений формулы I и/или в качестве антибиотиков. В еще одном своем аспекте по композиции настоящее изобретение относится к соединению формулы III или к его солям, где R1, R2, Р 1 и Р 2 определены в настоящем описании, и Р 4 представляет собой водород или карбоксизащитную группу, причем, указанные соединения могут быть использованы в качестве промежуточных соединений для получения соединений формулы I или II. В отдельных и различных своих аспектах по композиции настоящее изобретение также относится к соединениям формул 2, 5b, 7, 8, 10, 11 и 13, определенным в настоящем описании, или к их солям или к-2 007001 их защищенным производным, где указанные соединения могут быть использованы в качестве промежуточных соединений для получения соединений формулы I и/или в качестве антибиотиков. В других своих аспектах по композиции настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли. Не претендуя на какую-либо конкретную теорию, можно лишь указать, что соединения формулы I,очевидно, ингибируют биосинтез бактериальной клетки, и, тем самым, ингибируют рост бактерий или вызывают лизис бактерий. Поэтому, помимо других своих свойств, соединения формулы I обладают ценными свойствами, позволяющими использовать их в качестве антибиотиков. В соответствии с одним из своих аспектов по способу настоящее изобретение относится к способу лечения бактериальных инфекций у млекопитающих, где указанный способ предусматривает введение млекопитающему фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли. Кроме того, в другом своем аспекте по способу настоящее изобретение относится к способу ингибирования роста бактерий, где указанный способ предусматривает контактирование бактерий с ингибирующим рост количеством соединения формулы I или его фармацевтически приемлемой соли. В еще одном своем аспекте по способу настоящее изобретение относится к способу ингибирования биосинтеза клеточной стенки бактерий, где указанный способ предусматривает контактирование бактерий с определенным количеством соединения формулы I или его фармацевтически приемлемой соли,ингибирующим биосинтез клеточной стенки. Настоящее изобретение также относится к способу получения соединений формулы I или их соли. В соответствии с этим в другом своем аспекте настоящее изобретение относится к способу получения соединения формулы I или его соли, где указанный способ предусматривает:(a) осуществление взаимодействия гликопептида формулы 1, определенного в настоящем описании,с соединением формулы 2, определенным в настоящем описании; или(b) осуществление взаимодействия соединения формулы 10, определенного в настоящем описании,с соединением формулы 11, определенным в настоящем описании; или(c) осуществление взаимодействия соединения формулы 9, определенного в настоящем описании, с соединением формулы 13, определенным в настоящем описании,с получением соединения формулы I или его соли. В одном из предпочтительных вариантов осуществления изобретения вышеуказанный способ, кроме того, включает стадию получения фармацевтически приемлемой соли соединения формулы I. Настоящее изобретение также относится к продукту, полученному любым из этих способов. Настоящее изобретение также относится к соединению формулы I или к его фармацевтически приемлемой соли, предназначенным для использования в терапии. Кроме того, настоящее изобретение относится к использованию соединения формулы I или его фармацевтически приемлемой соли в целях приготовления лекарственных средств для лечения бактериальных инфекций у млекопитающих. Подробное описание изобретения Настоящее изобретение относится к новым соединениям гликопептида-цефалоспорина формулы I или к их фармацевтически приемлемым солям. Указанные соединения имеют множество хиральных центров, и поэтому эти соединения имеют указанную стереохимию. В частности, гликопептидная часть указанного соединения, очевидно, имеет стереохимию соответствующего природного гликопептида (то есть ванкомицина, хлорориентицина А и т.п.) Цефалоспориновая часть этой молекулы, очевидно, имеет стереохимию известных соединений цефалоспорина. Однако специалистам в данной области понятно, что в композициях по настоящему изобретению могут присутствовать небольшие количества изомеров,имеющих стереохимию, отличающуюся от указанной здесь стереохимии, при условии, что присутствие таких изомеров не будет значительно влиять на ценность данной композиции как единого целого. Кроме того, линкерная часть соединений по настоящему изобретению (то есть, R1) может содержать один или несколько хиральных центров. Обычно эта часть молекулы может быть получена в виде рацемической смеси. Однако, если необходимо, то могут быть использованы чистые стереоизомеры (то есть, отдельные энантиомеры или диастереомеры), либо может быть использована смесь, обогащенная каким-либо одним стереоизомером. Все указанные стереоизомеры и обогащенные смеси входят в объем настоящего изобретения. Кроме того, соединения по настоящему изобретению содержат несколько кислотных групп (то есть,групп карбоновой кислоты) и несколько основных групп (то есть, первичных аминовых и вторичных аминовых групп), и поэтому соединения формулы I могут существовать в различных солевых формах. Все такие солевые формы входят в объем настоящего изобретения. Кроме того, поскольку соединения формулы I содержат пиридиниевое кольцо, то может присутствовать, но необязательно, анионный противоион для пиридиниевой группы, включая, но не ограничиваясь ими, галогениды, такие как хлориды; карбоксилаты, такие как ацетат; и т.п.-3 007001 Кроме того, специалистам в данной области будет понятно, что в настоящее изобретение не входят лабильные или химически нестабильные соединения, которые не представляют какой-либо ценности изза своей нестабильности. Так, например, предпочтительные соединения формулы I содержат, по крайней мере, два атома углерода между двумя атомами кислорода (-O-), азота (-N) или серы (-S-) в -О-R1-N(R2)группе, поскольку в случае, если эти атомы будут разделены одним атомом углерода в полученном соединении (то есть, содержащем ацетальную, полуацетальную, кетальную, полукетальную, аминальную,полуаминальную или тиокетальную группу и т.п.), то оно может оказаться гидролитически нестабильным под действием кислот. Предпочтительные варианты осуществления изобретения В соединениях формулы I присутствуют следующие предпочтительные заместители и радикалы. В предпочтительном варианте осуществления настоящее изобретение относится к соединениям формулы Iа или к их фармацевтически приемлемым солям, где R1, R2, R3 и m определены в настоящем описании,включая предпочтительные варианты. В предпочтительном варианте осуществления изобретения R1 представляет собой -Ya-Yb-, то есть,когда n равно 0. В этом варианте Ya и Yb независимо представляют собой С 1-5 алкиленовые группы. Предпочтительно Ya и Yb независимо выбраны из C1-3 алкилена и более предпочтительно С 1-2 алкилена. Более предпочтительно, когда Ya и Yb, взятые вместе (то есть R1) образуют группу -(СН 2)2-8. Еще более предпочтительно, когда Ya и Yb, взятые вместе, образуют группы -(СН 2)2-, -(СН 2)3-, -(СН 2)4-, (СН 2)5 или -(СН 2)6. В особенно предпочтительном варианте по настоящему изобретению Ya и Yb, взятые вместе, образуют группу -(СН 2)3-. В другом предпочтительном варианте изобретения R1 представляет собой -Ya-WYb-, то есть, когда n равно 1. В этом варианте Ya и Yb независимо представляют собой С 1-5 алкилен. Если Ya или Yb представляют собой алкиленовую группу, то такой алкиленовой группой предпочтительно является C1-3 алкиленовая группа, более предпочтительно С 1-2 алкиленовая группа и еще более предпочтительно группа -(CH2)1-2. В особенно предпочтительном варианте изобретения оба Ya и Yb представляют собой -СН 2- и W представляет собой С 6-10 арилен и более предпочтительно W представляет собой фенилен. В другом предпочтительном варианте изобретения оба Ya и Yb представляют собойW, если он присутствует, предпочтительно представляет собой С 6-10 арилен или -О-. Более предпочтительно W представляет собой фенилен или -О-. Предпочтительно R2 представляет собой водород или С 1-3 алкил. Более предпочтительно R2 представляет собой водород. Предпочтительно каждый R3, если он присутствует, независимо выбран из C1-6 алкила, С 3-6 циклоалкила, -ORd, -SRd, -F или -Сl, либо две смежные группы R3, взятые вместе, образуют С 3-6 алкилен. В предпочтительном варианте осуществления изобретения R4 представляет собой гидрокси и R5 представляет собой водород. Предпочтительно R6 представляет собой водород и R7 представляет собой метил. Предпочтительно R8 представляет собой водород.-4 007001 Предпочтительно, один из X1 и X2 представляет собой хлор, а другой - водород либо оба они представляют собой хлор. Более предпочтительно, если оба X1 и X2 представляют собой хлор. В предпочтительном варианте изобретения R4 представляет собой гидрокси; R5 представляет собой водород; R6 представляет собой водород; R7 представляет собой метил; R8 представляет собой водород и оба X1 и X2 представляют собой хлор (то есть гликопептидная часть представляет собой ванкомицин). В другом предпочтительном варианте изобретения R4 представляет собой водород; R5 представляет собой гидрокси; R6 представляет собой водород; R7 представляет собой метил; R8 представляет собой группу формулы (i) и оба X1 и X2 представляют собой хлор (то есть гликопептидная часть представляет собой хлорориентицин или А 82846 В). В предпочтительном варианте изобретения m равно 0. В другом предпочтительном варианте изобретения m равно 1 или 2 и более предпочтительно 1. В еще одном предпочтительном варианте изобретения m равно 2, а две группы R3, взятые вместе, образуют С 3-5 алкиленовую группу и более предпочтительно С 3-4 алкиленовую группу. Предпочтительной группой соединений формулы I являются соединения формулы Iа, где R1 представляет собой -Ya-(W)n-Yb-, где n равно 0, а Ya и Yb, взятые вместе, образуют группу -(СН 2)2-8, R2 представляет собой водород и m равно 0, или их фармацевтически приемлемые соли. В этом варианте изобретения R1 (то есть, Yа и Yb, взятые вместе) предпочтительно представляют собой группы -(СН 2)2, -(СН 2)3-,-(СН 2)4-, -(СН 2)5 или -(СН 2)6, и более предпочтительно -(СН 2)3-. Другой предпочтительной группой соединений формулы I являются соединения формулы Iа, гдеR1, R2, R3 и m определены в табл. 1, или их фармацевтически приемлемые соли. Таблица 1 В промежуточном соединении формулы IIQ предпочтительно представляет собой галоген или определенную пиридиниевую группу. Р 1 предпочтительно представляет собой водород или трет-бутоксикарбонил. Р 2 предпочтительно представляет собой водород или трифенилметил. Р 3 предпочтительно представляет собой водород или п-метоксибензил.R1, R2, R3 и m предпочтительно являются такими, как они были определены в настоящем описании,включая любые предпочтительные варианты, заместители или группы. В промежуточном соединении формулы III Р 1 предпочтительно представляет собой водород или трет-бутоксикарбонил. Р 2 предпочтительно представляет собой водород, формил или трифенилметил. Р 4 предпочтительно представляет собой водород, С 1-4 алкил или п-метоксибензил.R1 и R2 предпочтительно являются такими, как они были определены в настоящем описании, включая любые предпочтительные варианты изобретения, заместители или группы. Определения При описании соединений, композиций, методов и способов по настоящему изобретению используются нижеследующие термины, которые имеют значения, определенные ниже, если это не оговорено особо. Термин "алкил" означает одновалентную насыщенную углеводородную группу, которая может быть прямой или разветвленной. Если это не оговорено особо, то такие алкильные группы обычно содержат от 1 до 10 атомов углерода. Характерными алкильными группами являются, например, метил,этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил, ноктил, н-нонил, н-децил и т.п. Термин "алкилен" означает двухвалентную насыщенную углеводородную группу, которая может быть прямой или разветвленной. Если это не оговорено особо, то такие алкиленовые группы обычно содержат от 1 до 10 атомов углерода. Характерными алкиленовыми группами являются, например, метилен, этан-1,2-диил ("этилен"), пропан-1,2-диил, пропан-1,3-диил, бутан-1,4-диил, пентан-1,5-диил и т.п. Термин "алкенил" означает одновалентную ненасыщенную углеводородную группу, которая может быть прямой или разветвленной и которая имеет по крайней мере одну, а обычно 1, 2 или 3 углеродуглеродных двойных связей. Если это не оговорено особо, то такие алкенильные группы обычно содержат от 2 до 10 атомов углерода. Характерными алкенильными группами являются, например, этенил, нпропенил, изопропенил, н-бут-2-енил, н-гекс-3-енил и т.п. Термин "алкинил" означает одновалентную ненасыщенную углеводородную группу, которая может быть прямой или разветвленной и которая имеет по крайней мере одну, а обычно 1, 2 или 3 углеродуглеродных тройных связей. Если это не оговорено особо, то такие алкинильные группы обычно содержат от 2 до 10 атомов углерода. Характерными алкенильными группами являются, например, этинил, нпропинил, н-бут-2-инил, н-гекс-3-инил и т.п. Термин "арил" означает одновалентный ароматический углеводород, имеющий одно кольцо (то есть фенил) или конденсированные кольца (то есть нафталин). Если это не оговорено особо, то такие арильные группы обычно содержат от 6 до 10 атомов углерода в кольце. Характерными арильными группами являются, например, фенил, нафталин-1-ил, нафталин-2-ил и т.п. Термин "арилен" означает двухвалентный ароматический углеводород, имеющий одно кольцо (то есть фенилен) или конденсированные кольца (то есть нафталиндиил). Если это не оговорено особо, то такие ариленовые группы обычно содержат от 6 до 10 атомов углерода в кольце. Характерными ариленовыми группами являются, например, 1,2-фенилен, 1,3-фенилен, 1,4-фенилен, нафталин-1,5-диил, нафталин-2,7-диил и т.п.-6 007001 Термин "циклоалкил" означает одновалентную насыщенную карбоциклическую углеводородную группу. Если это не оговорено особо, то такие циклоалкильные группы обычно содержат от 3 до 10 атомов углерода. Характерными циклоалкильными группами являются, например, циклопропил, циклобутил, циклопентил, циклогексил и т.п. Термин "циклоалкилен" означает двухвалентную насыщенную карбоциклическую углеводородную группу. Если это не оговорено особо, то такие циклоалкильные группы обычно содержат от 3 до 10 атомов углерода. Характерными циклоалкиленовыми группами являются, например, циклопропан-1,2-диил,циклобутил-1,2-диил, циклобутил-1,3-диил, циклопентил-1,2-диил, циклопентил-1,3-диил, циклогексил 1,2-диил, циклогексил-1,3-диил, циклогексил-1,4-диил и т.п. Термин "галоген" означает фтор, хлор, бром и йод. Термин "гетероарил" означает одновалентную ароматическую группу, имеющую одно кольцо или два конденсированных кольца и содержащую в кольце, по крайней мере, один гетероатом (обычно 1-3 гетероатома), выбранный из азота, кислорода или серы. Если это не оговорено особо, то такие гетероарильные группы обычно содержат всего от 5 до 10 атомов углерода в кольце. Характерными гетероарильными группами являются, например, одновалентные группы, такие как пиррол, имидазол, тиазол,оксазол, фуран, тиофен, триазол, пиразол, изоксазол, изотиазол, пиридин, пиразин, пиридазин, пиримидин, триазин, индол, бензофуран, бензотиофен, бензимидазол, бензотиазол, хинолин, изохинолин, хиназолин, хиноксалин и т.п., где положение присоединения может быть у любого доступного атома углерода или азота в кольце. Термин "гетероарилен" означает двухвалентную ароматическую группу, имеющую одно кольцо или два конденсированных кольца и содержащую в кольце, по крайней мере, один гетероатом (обычно 13 гетероатома), выбранный из азота, кислорода или серы. Если это не оговорено особо, то такие гетероариленовые группы обычно содержат всего от 5 до 10 атомов в кольце. Характерными гетероариленовыми группами являются, например, двухвалентные группы, такие как пиррол, имидазол, тиазол, оксазол,фуран, тиофен, триазол, пиразол, изоксазол, изотиазол, пиридин, пиразин, пиридазин, пиримидин, триазин, индол, бензофуран, бензотиофен, бензимидазол, бензотиазол, хинолин, изохинолин, хиназолин, хиноксалин и т.п., где положение присоединения может быть у любого доступного атома углерода или азота в кольце. Термин "гетероциклил" или "гетероцикл" означает одновалентную насыщенную или ненасыщенную (неароматическую) группу, имеющую одно кольцо или множество конденсированных колец и содержащую в кольце по крайней мере один гетероатом (обычно 1-3 гетероатома), выбранный из азота,кислорода или серы. Если это не оговорено особо, то такие гетероциклические группы обычно содержат всего от 2 до 9 атомов в кольце. Характерными гетероциклическими группами являются, например, одновалентные группы, такие как пирролидин, имидазолидин, пиразолидин, пиперидин, 1,4-диоксан, морфолин, тиоморфолин, пиперазин, 3-пирролин и т.п., где положение присоединения может быть у любого доступного атома углерода или азота в кольце. Используемый здесь термин "цефалоспорин" известен специалистам как -лактамовая циклическая система, имеющая нижеследующую общую формулу и систему нумерации: Используемый здесь термин "гликопептидный антибиотик" или "гликопептид" известен специалистам как антибиотик класса гликопептидов или далбапептидов. См., например, R. Nagarajan, "Glycopeptide Antibiotics", Marcel Dekker, Inc. (1994) и цитируемые там работы. Характерными гликопептидами являются ванкомицин, А 82846 А (эремомицин), А 82846 В (хлорориентицин А), А 82846 С, РА-42867-А(ориентицин А), РА-42867-С, PA-42867-D и т.п. Используемый здесь термин "ванкомицин" известен специалистам как гликопептидный антибиотик под названием ванкомицин. В соединениях по настоящему изобретению положение присоединения линкерной группы находится у "С-конца" ванкомицина. Термин "фармацевтически приемлемая соль" означает соль, которая является приемлемой для введения пациенту, такому как млекопитающее (например, соли, являющиеся безопасными для млекопитающего при введении в соответствии с данной схемой приема лекарственного средства). Такие соли могут быть получены из фармацевтически приемлемых неорганических или органических оснований и из фармацевтически приемлемых неорганических или органических кислот. Солями, полученными из фармацевтически приемлемых неорганических оснований, являются соли алюминия, аммония, кальция, меди, железа(3), железа(2), лития, магния, марганца(3), марганца(2), калия, натрия, цинка и т.п. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Солями, полученными из фармацевтически приемлемых органических оснований, являются соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, природные амины и т.п., такие как аргинин, бетаин, кофеин, хо-7 007001 лин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин,этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперадин, полиаминовые смолы, прокаин,пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Солями, полученными из фармацевтически приемлемых кислот, являются соли уксусной, аскорбиновой, бензолсульфоновой, бензойной, камфосульфоновой, лимонной, этансульфоновой, фумаровой, глюконовой, глюкуроновой, глутаминовой, гиппуровой, бромисто-водородной, хлористо-водородной, изетионовой, молочной, лактобионовой, малеиновой, яблочной, миндальной, метансульфоновой, слизевой, нафталинсульфоновой, никотиновой, азотной, памовой, пантотеновой, фосфорной, янтарной, серной, винной, п-толуолсульфоновой кислоты и т.п. Особенно предпочтительными являются лимонная, бромисто-водородная, хлористо-водородная,малеиновая, фосфорная, серная и винная кислоты. Термин "его соль" относится к соединению, образованному в случае, когда атом водорода кислоты заменен катионом, таким как катион металла или органический катион и т.п. (например, катион NH4+ и т.п.). Предпочтительно, чтобы данная соль была фармацевтически приемлемой солью, хотя это требование является необязательным для солей промежуточных соединений, которые не предназначены для введения пациенту. Термин "терапевтически эффективное количество" означает количество, достаточное для эффективного лечения при его введении пациенту, нуждающемуся в таком лечении. Используемый здесь термин "лечение" или "терапия" означает лечение или терапию заболевания или клинического состояния (такого как бактериальная инфекция) у пациента, такого как млекопитающее (в частности, человека или животного-компаньона), где указанный термин включает:(a) предупреждение заболевания или клинического состояния, которое может возникнуть, то есть,профилактическое лечение пациента;(b) ослабление симптомов данного заболевания или клинического состояния, то есть ликвидацию данного заболевания или клинического состояния или стимуляцию его ремиссии у пациента,(c) подавление развития заболевания или клинического состояния, то есть, замедление или прекращение развития такого заболевания или клинического состояния у пациента, или(d) ослабление симптомов данного заболевания или клинического состояния у пациента. Термин "ингибирующее рост количество" означает количество, достаточное для ингибирования роста или размножения микроорганизмов, или количество, достаточное для инициации гибели или лизиса указанных микроорганизмов, включая грамположительные бактерии. Термин "количество, ингибирующее биосинтез клеточной стенки", означает количество, достаточное для ингибирования биосинтеза клеточной стенки микроорганизмов, включая грамположительные бактерии. Термин "удаляемая группа" означает функциональную группу или атом, которые могут быть заменены другой функциональной группой или атомом в реакции замещения, такой как реакция нуклеофильного замещения. Так, например, характерными удаляемыми группами являются хлор, бром и йод; и сложноэфирные группы сульфоновой кислоты, такие как мезилат, тозилат, брозилат, нозилат и т.п.; группы активированного сложного эфира, такие как 7-азабензотриазол-1-окси и т.п.; ацилоксигруппы,такие как ацетокси, трифторацетокси и т.п. Термин "его защищенные производные" означает производные конкретного соединения, в котором одна или несколько функциональных групп указанного соединения защищены от нежелательных реакций защитными или блокирующими группами. Функциональными группами, которые могут быть защищены, являются, например, группы карбоновых кислот, аминогруппы, гидроксильные группы, тиоловые группы, карбонильные группы и т.п. Характерными защитными группами для карбоновых кислот являются сложные эфиры (такие как п-метоксибензиловый эфир), амиды и гидразиды; для аминогрупп - карбаматы (такие как трет-бутоксикарбонил) и амиды; для гидроксильных групп - простые эфиры и сложные эфиры; для тиоловых групп - простые тиоэфиры и сложные тиоэфиры; для карбонильных групп ацетали и кетали и т.п. Такие защитные группы хорошо известны специалистам и описаны, например, вT.W. GreeneG.M. Wuts, Protective Groups in Organic Synthesis, Third Edition., Wiley, New York, 1999 и в цитируемых там работах. Термин "аминозащитная группа" означает защитную группу, подходящую для предотвращения нежелательных реакций аминогруппы. Характерными аминозащитными группами являются, но не ограничиваются ими, трет-бутоксикарбонил (ВОС), тритил (Тr), бензилоксикарбонил (Cbz), 9-флуоренилметоксикарбонил (Fmoc), формил, триметилсилил (TMS), трет-бутилдиметилсилил (TBS) и т.п. Термин "карбоксизащитная группа" означает защитную группу, подходящую для предотвращения нежелательных реакций у карбоксигруппы. Характерными карбоксизащитными группами являются, но ими не ограничиваются, сложные эфиры, такие как метиловый, этиловый, трет-бутиловый, бензиловый-8 007001 Общие методы синтеза Перекрестно-сшитые соединения гликопептида-цефалоспорина по настоящему изобретению могут быть получены из легко доступных исходных материалов в соответствии с общими методами и методами, описанными ниже. При этом следует отметить, что хотя далее приведены типичные или предпочтительные условия реакций (то есть, температура и время реакции, молярные соотношения реагентов, растворители, давление и т.п.), однако, могут быть также использованы и другие условия реакции, если это не оговорено особо. Оптимальные условия реакции могут варьироваться в зависимости от конкретно используемых реагентов или растворителей, и такие условия могут быть легко определены любым специалистом с помощью обычных методов по оптимизации. Кроме того, для каждого специалиста очевидно, что для предохранения некоторых функциональных групп от нежелательных реакций необходимо или желательно использовать стандартные защитные группы. Выбор подходящей защитной группы для конкретной функциональной группы, а также подходящих условий для защиты таких функциональных групп и снятия защиты с этих функциональных групп может быть осуществлен любым специалистом. Если это необходимо, то могут быть использованы и другие защитные группы, которые не указаны в описанных здесь способах. Так, например, множество защитных групп и способы их введения и удаления описаны в T.W. GreeneG.M. Wuts, ProtectingGroups in Organic Synthesis, Third Edition., Wiley, New York, 1999 и в цитируемых там работах. В предпочтительном методе синтеза соединение формулы I или его соль получают путем взаимодействия гликопептида формулы 1 где R4, R5, R6, R7, R8, X1 и X2 определены в настоящем описании, с соединением формулы 2 или с его солью или защищенным производным где R1, R2, R3 и m определены в настоящем описании, с получением соединения формулы I или его соли или защищенного производного. Указанную реакцию обычно осуществляют посредством взаимодействия гликопептида 1 или его соли примерно с 0,5-1,5 эквивалентами и предпочтительно примерно с 0,9-1,1 эквивалентами соединения формулы 2 в инертном разбавителе,"таком как ДМФ, с использованием стандартного реагента сочетания"карбоновая кислота - амин (пептид)". В этой реакции обычно гликопептид 1 или его соль сначала подвергают взаимодействию с реагентом сочетания в присутствии избытка амина, предпочтительно примерно с 1,8-2,2 эквивалентами амина, такого как диизопропилэтиламин, при температуре примерно от-20 до 25 С, предпочтительно при комнатной температуре в течение примерно 0,25-3 ч. Затем добавляют предпочтительно избыток трифторуксусной кислоты (обычно примерно 2 эквивалента) с получениемTFA-соли любого избытка диизопропилэтиламина. Затем реакционную смесь обычно охлаждают до температуры примерно от -20 до 10 С и предпочтительно примерно до 0 С и добавляют промежуточное соединение 2, после чего добавляют избыточное количество 2,4,6-коллидина. Эту реакционную смесь обычно выдерживают примерно при 0 С в течение примерно 1-6 ч или до тех пор, пока реакция не будет в основном завершена. Предпочтительный реагент сочетания, используемый в данной реакции, содержит примерно 0,5-1,5 эквивалентов, предпочтительно, примерно 0,9-1,1 эквивалентов гексафторфосфата бензотриазол-1 илокситрипирролидинфосфония (РуВОР) и примерно 0,5-1,5 эквивалентов, предпочтительно, примерно 0,9-1,1 эквивалентов 1-гидроксибензотриазола (НОВТ) или 1-гидрокси-7-азабензотриазола (НОАТ). Другими подходящими реагентами сочетания являются гексафторфосфат О-(7-азабензотриазол-1-ил)-9 007001(ВОР-С 1), дифенилфосфорилазид (DPPA), хлорангидрид дифенилфосфиновой кислоты, дифенилхлорфосфат (DPCP) и НОАТ, пентафторфенилдифенилфосфинат и т.п. После завершения реакции сочетания любую защитную группу, присутствующую в этом продукте,удаляют с использованием обычных методов и реагентов. После завершения реакции продукты реакции,то есть соединение формулы I, выделяют и очищают с использованием обычных методов, таких как колоночная хроматография, ВЭЖХ, перекристаллизации и т.п. Гликопептиды формулы 1, подходящие для использования в вышеописанном методе, либо являются коммерчески доступными, либо они могут быть получены путем ферментации соответствующего гликопептид-продуцирующего микроорганизма с последующим выделением указанного гликопептида из полученной сбраживаемой среды с использованием обычных методов и оборудования. Промежуточное соединение цефалоспорина 2, используемое в вышеописанном методе, может быть легко получено из коммерчески доступных исходных материалов и реагентов с использованием обычных методов. Так, например, промежуточное соединение 2 может быть получено, как показано на схеме А. Схема А Как проиллюстрировано на схеме А, тиазоловое промежуточное соединение 3 (где R9 представляет собой аминозащитную группу, такую как тритильная группа, и R10 представляет собой карбоксизащитную группу, такую как этильная группа) сначала подвергают взаимодействию с -функционализированным амином формулы 4 (где R1 и R2 определены в настоящем описании, R11 представляет собой аминозащитную группу, такую как трет-бутоксикарбонильную (ВОС) группу и Z1 представляет собой удаляемую группу, такую как хлор, бром, йод, мезилат, тозилат и т.п.) с получением после удаления карбоксизащитной группы (то есть R10) промежуточного соединения формулы 5 а. Указанную реакцию обычно осуществляют сначала путем взаимодействия соединения 3 примерно с 1,0-1,1 эквивалентами, предпочтительно примерно с 1,02-1,06 эквивалентами соединения формулы 4 в инертном разбавителе, таком как ДМФ, при температуре примерно от 0 до 50 С и предпочтительно при комнатной температуре, в течение примерно 0,5-6 ч или до тех пор, пока реакция не будет в основном завершена. Эту реакцию обычно проводят в присутствии избыточного количества предпочтительно примерно 1,1-5 эквивалентов основания, такого как карбонат цезия. Кроме того, если Z1 представляет собой хлор или бром, то для ускорения реакции при образовании йодного производного 4 in situ, добавляют, но необязательно, каталитическое количество предпочтительно примерно 0,2-0,5 эквивалентов йодида триалкиламмония, такого как йодид тетрабутиламмония. Затем, после удаления карбоксизащитной группы (то есть R10) получают промежуточное соединение 5 а. Так, например, если карбоксизащитной группой является алкиловый сложный эфир, такой как этильная группа, то этот сложный эфир легко гидролизуется до карбоновой кислоты при взаимодействии- 10007001 указанного сложного эфира с избыточным количеством предпочтительно примерно с 1,1-2,5 эквивалентами гидроксида щелочного металла, такого как гидроксид натрия или гидроксид калия. Эту реакцию обычно проводят в инертном разбавителе, таком как этанол, при температуре примерно от 0 до 100 С в течение примерно 0,5-6 ч или до тех пор, пока реакция не будет в основном завершена, в результате чего получают промежуточное соединение 5 а. Тиазоловые соединения формулы 3 либо являются коммерчески доступными, например продукт фирмы Aldrich Milwaukee, WI, либо они могут быть получены из коммерчески доступных исходных материалов и реагентов с использованием стандартных методов. Аналогичным образом, -функционализированные амины формулы 4 могут быть легко получены из коммерчески доступных исходных материалов и реагентов с использованием стандартных методов. Предпочтительными соединениями формулы 4 являются, например, N-ВОС-3-бромпропиламин, N-BOC6-йодгексиламин, N-BOC-2-(2-йодэтокси)этиламин; N-BOC-4-(2-йодметил)бензиламин и т.п. Указанные соединения могут быть легко получены из коммерчески доступных исходных материалов с использованием хорошо известных реагентов и стандартных реакционных условий. Затем промежуточное соединение 5 а хлорируют и получают промежуточное соединение 5b. Эту реакцию обычно осуществляют путем взаимодействия соединения 5 а примерно с 1,0-1,2 эквивалентами хлорирующего агента, такого как N-хлорсукцинимид, в инертном разбавителе, таком как хлороформ или ДМФ, при комнатной температуре в течение примерно 6-24 ч или до тех пор, пока реакция не будет в основном завершена. Затем 5-хлор-1,3-тиазоловое промежуточное соединение 5b подвергают реакции сочетания с промежуточным соединением 6 (где R12 представляет собой водород или подходящую карбоксизащитную группу, такую как п-метоксибензильная группа) с получением промежуточного соединения 7. Если R12 представляет собой п-метоксибензил, то промежуточное соединение 6 является коммерчески доступным соединением, поставляемым фирмой Otsuka, Japan. Обычно эту реакцию осуществляют путем взаимодействия соединения 5b примерно с 0,8-1 эквивалентами соединения 6 в присутствии реагента сочетания в стандартных условиях, обычно используемых для реакции сочетания. Предпочтительным реагентом сочетания для этой реакции является оксихлорид фосфора (обычно примерно 1,1-1,2 эквивалента) и избыточное количество амина, такого как 2,4,6-коллидин или диизопропилэтиламин. Такую реакцию сочетания обычно проводят в инертном разбавителе, таком как ТГФ, при температуре примерно от -50 до 25 С в течение примерно 0,5-6 ч или до тех пор, пока реакция не будет в основном завершена, в результате чего получают промежуточное соединение 7. Во избежание изомеризации эту реакцию предпочтительно проводят при -35 С с использованием 2,4,6-коллидина в качестве основания. Затем промежуточное соединение 7 подвергают взаимодействию с пиридином или с замещенным пиридином с получением промежуточного соединения 8, где R3 и n определены в настоящем описании. Обычно эту реакцию сначала проводят путем обмена хлора в соединении 7 на йод путем взаимодействия соединения 7 примерно с одним эквивалентом йодида натрия в ацетоне (реакция Финкельштейна) или в ДМФ при комнатной температуре в течение примерно 0,25-2 ч. Полученное йодное промежуточное соединение обычно не выделяют, и оно реагирует in situ примерно с 1,1-1,6 эквивалентами пиридина или замещенного пиридина с образованием соединения 8. Обычно эту реакцию осуществляют при комнатной температуре в течение примерно 1-12 ч или до тех пор, пока реакция не будет в основном завершена. Пиридин или замещенные пиридины, используемые в этой реакции, либо являются коммерчески доступными, либо они могут быть получены из коммерчески доступных исходных материалов и реагентов с использованием обычных методов. Характерными пиридиновыми производными, используемыми в этой реакции, являются пиридин, 2-пиколин, 3-пиколин, 4-пиколин, 2-метоксипиридин, 3-метоксипиридин, 4 метоксипиридин, 2-тиометоксипиридин, 3-тиометоксипиридин, 4-тиометоксипиридин, 4-карбокситиометоксипиридин, 2-фторпиридин, 3-фторпиридин, 4-фторпиридин, 2-хлорпиридин, 3-хлорпиридин, 4 хлорпиридин, 2-фенилпиридин, 3-фенилпиридин, 4-фенилпиридин, 4-циклопропилпиридин, никотиновая кислота, изоникотиновая кислота, никотинамид, изоникотинамид, 2,3-лутидин, 3,4-лутидин, 3,5-лутидин,3,4-диметоксипиридин, 4-метокси-3-метилпиридин, 4-фтор-3-метоксипиридин, 2,3-циклопентенопиридин, 2,3-циклогексенопиридин и т.п. Альтернативно, промежуточное соединение 5b может быть подвергнуто реакции сочетания с соединением формулы 9 где R3, R12 и m определены в настоящем описании, с получением промежуточного соединения 8. Эту реакцию обычно проводят путем взаимодействия соединения 5b примерно с 0,9-1,1 эквивалентами промежуточного соединения 9 или с его солью в инертном растворителе, таком как ДМФ, в присутствии реагента сочетания, такого как 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC); РуВОР и НОАТ или НОВТ; HATU; BOP-Cl; DPPA; DPCP и НОАТ и т.п. Обычно реакцию сочетания проводят при тем- 11007001 пературе примерно от -40 до 25 С в течение примерно 1-12 ч или до тех пор, пока реакция не будет в основном завершена. Соединения формулы 9 могут быть легко получены из промежуточного соединения 6 путем взаимодействия соединения 6 с пиридином или с замещенным пиридином в реакционных условиях, аналогичных описанным выше. Затем, после удаления защитных групп из промежуточного соединения 8 с использованием стандартных методов и реагентов, получают цефалоспориновое промежуточное соединение 2. Так, например,когда R9 представляет собой тритил, R11 представляет собой трет-бутоксикарбонил и R12 представляет собой пара-метоксибензил, указанные защитные группы обычно удаляют путем обработки соединения 8 избытком трифторуксусной кислоты и избытком анизола или триэтилсилана в инертном разбавителе,таком как дихлорметан или гептан при комнатной температуре, в течение примерно 1-12 ч или до тех пор, пока реакция не завершится. После удаления защитной группы полученный цефалоспорин 2 обычно выделяют и очищают с использованием стандартных способов, таких как осаждение, лиофилизация и обращенно-фазовая ВЭЖХ. Альтернативно, соединения формулы I могут быть получены посредством взаимодействия гликопептидного производного формулы 10 или его соли, где R1, R2, R4, R5, R6, R7, R8, X1 и X2 определены в настоящем описании, с производным цефалоспорина формулы 11 или с его солью или защищенным производным (где R3 и m определены в настоящем описании) с получением соединения формулы I или его соли. Указанную реакцию обычно осуществляют путем взаимодействия соединения 10 примерно с 1-1,5 эквивалентами соединения 11 в инертном разбавителе, таком как вода, метанол или их смеси, при рН примерно от 4 до 6,5. Эту реакцию обычно проводят при температуре примерно от -20 до 40 С в течение примерно 1-6 ч или до тех пор, пока реакция не будет в основном завершена. Производные ванкомицина формулы 10, используемые в данной реакции, могут быть легко получены посредством реакции сочетания ванкомицина или его соли с фталимидным производным формулы где R1 определен в настоящем описании, в стандартных условиях реакции сочетания. Так, например, эту реакцию обычно осуществляют путем взаимодействия ванкомицина примерно с 1,1-1,2 эквивалентами указанного фталимидного соединения в присутствии реагента сочетания, такого как РуВОР и НОАТ и т.п. и подходящего основания, такого как диизопропилэтиламин. Обычно эту реакцию проводят в инертном разбавителе, таком как ДМФ, при температуре примерно от -20 до 40 С в течение примерно 0,5-6 ч- 12007001 или до тех пор, пока реакция не будет в основном завершена. Фталимидные соединения, используемые в данной реакции, могут быть легко получены с использованием обычных методов и реагентов. Производные цефалоспорина формулы 11 могут быть получены, например, посредством реакции сочетания соединения вышеуказанной формулы 9 с тиазоловым соединением формулы 12 или с его солью, где R13 представляет собой водород или аминозащитную группу (такую как формильная или тритильная группа). Эту реакцию сочетания обычно осуществляют путем взаимодействия соединения 9 примерно с 0,9-1,1 эквивалентами соединения 12 в инертном разбавителе, таком как ДМФ, в присутствии реагента сочетания, такого как EDC и НОАТ, и подходящего основания, такого как 2,4,6 коллидин. Обычно эту реакцию проводят при температуре примерно от -20 до 20 С в течение примерно 0,5-6 ч или до тех пор, пока реакция не будет в основном завершена. Кроме того, соединения формулы I могут быть получены посредством взаимодействия гликопептидного производного формулы 13 или его соли, где R1, R2, R4, R5, R6, R7, R8, X1 и X2 определены в настоящем описании, с соединением вышеуказанной формулы 9 с получением соединения формулы I или его соли. Указанную реакцию сочетания обычно осуществляют путем взаимодействия соединения 13 с реагентом сочетания, таким как DIPC и НОАТ, и примерно с 0,5-2 эквивалентами соединения 9 в инертном разбавителе, таком как ДМФ, в присутствии подходящего основания, такого как 2,4,6-коллидин. Эту реакцию обычно осуществляют при температуре примерно от -20 до 40 С в течение примерно 1-6 ч или до тех пор, пока реакция не будет в основном завершена. Соединение формулы 13, используемое в данной реакции, может быть легко получено посредством реакции сочетания ванкомицина с вышеописанным промежуточным соединением 5b с использованием обычных способов сочетания, описанных в настоящей заявке. Более подробное описание конкретных реакционных условий и способов получения характерных соединений по настоящему изобретению или их промежуточных соединений представлено в нижеследующих примерах. Фармацевтические композиции Перекрестно-сшитые соединения гликопептида-цефалоспорина по настоящему изобретению обычно вводят пациенту в виде фармацевтической композиции. В соответствии с этим, в одном из своих аспектов по композиции, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель или наполнитель и терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли. В фармацевтической композиции по настоящему изобретению может быть использован любой подходящий носитель или наполнитель. Выбор конкретного носителя или наполнителя или комбинаций носителей или наполнителей зависит от способа введения, используемого для лечения конкретного пациента, или для ликвидации бактериальной инфекции конкретного типа. В соответствии с этим, получение- 13007001 фармацевтической композиции, подходящей для конкретного способа введения, такого как пероральное введение, местное применение, введение путем ингаляции или парентеральное введение, может быть с успехом осуществлено самим специалистом-фармацевтом. Кроме того, ингредиенты для таких композиций являются коммерчески доступными и поставляются, например, фирмой Sigma, P.O. Box 14508, St.Louis, МО 63178. Кроме того, методы получения стандартных фармацевтических композиций подробно описаны в руководстве Remington's Pharmaceutical Sciences, Mace Publishing. Co., Philadelphia, PA 17th Ed.(1985) и в руководстве "Modern Pharmaceutics", Marcel Dekker, Inc. 3th Ed. (G.S. BankerC.T. Rhodes,Eds.). Фармацевтические композиции по настоящему изобретению обычно содержат терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли. Обычно такие фармацевтические композиции содержат примерно от 0,1 до 90 мас.% активного агента и, в основном,примерно от 10 до 30% активного агента. Предпочтительные фармацевтические композиции по настоящему изобретению являются подходящими для парентерального введения и, в частности, для внутривенного введения. Такие фармацевтические композиции обычно содержат стерильный физиологически приемлемый водный раствор, содержащий терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли. Физиологически приемлемые водные растворы носителя, подходящие для внутривенного введения активных агентов, хорошо известны специалистам. Такими водными растворами являются, например,5% декстроза, растворы Рингера (лактат-содержащий раствор Рингера для инъекций, лактат-содержащий раствор Рингера + 5% декстроза для инъекций, ацилированный раствор Рингера для инъекций), нормозол-М, изолит Е и т.п. Такие водные растворы могут содержать, но необязательно, сорастворитель, например полиэтиленгликоль, хелатообразующий агент, например этилендиаминтетрауксусную кислоту; солюбилизирующий агент, например, циклодекстрин; антиоксидант, например метабисульфит натрия и т.п. Если это необходимо, то водные фармацевтические композиции по настоящему изобретению могут быть лиофилизованы, а затем, перед их введением, они могут быть разбавлены подходящим носителем. В предпочтительном варианте осуществления изобретения фармацевтическая композиция представляет собой лиофилизованную композицию, содержащую фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли. Носитель в данной композиции, предпочтительно, содержит сахарозу, маннит, декстрозу, декстран, лактозу или их комбинацию. Более предпочтительно, указанный носитель содержит сахарозу, маннит или их комбинацию. В одном из вариантов осуществления изобретения фармацевтические композиции по настоящему изобретению содержат циклодекстрин. Предпочтительным циклодекстрином, используемым в фармацевтических композициях по настоящему изобретению, является гидроксипропилциклодекстрин или сульфобутиловый эфир -циклодекстрина. В указанных композициях циклодекстрин составляет примерно 1-25 мас.% и предпочтительно примерно 2-10 мас.% данной композиции. Кроме того, массовое отношение циклодекстрина к активному агенту обычно составляет примерно от 1:1 до 10:1. Фармацевтические композиции по настоящему изобретению предпочтительно изготавливают в виде стандартной лекарственной формы. Термин "стандартная лекарственная форма" означает физически дискретную форму, подходящую для введения определенной дозы пациенту, то есть каждая такая форма содержит предварительно определенное количество активного агента, рассчитанное для продуцирования нужного терапевтического эффекта указанной лекарственной формой, взятой отдельно или в комбинации с одним или несколькими другими лекарственными формами. Так, например, указанные стандартные лекарственные формы могут быть упакованы в стерильные герметично запаянные ампулы и т.п. Ниже проиллюстрировано приготовление характерных фармацевтических композиций по настоящему изобретению. Пример приготовления композиции А. Замороженный раствор, подходящий для получения раствора для инъекций, получали следующим способом. Общая методика. Наполнители, если они используются, растворяют примерно в 80% воды для инъекций, добавляют активное соединение и растворяют. рН доводят до 3-4,5 добавлением 1 М гидроксида натрия, а затем объем доводят до 95% от конечного объема путем добавления воды для инъекций. рН контролируют и корректируют, если это необходимо, и объем доводят до конечного объема добавлением воды для инъекций. Затем полученную композицию фильтруют в стерильных условиях через фильтр- 14007001 0,22 мк и помещают в стерильный сосуд в условиях асептики. Сосуд закрывают крышкой, помечают и хранят в замороженном виде. Пример приготовления композиции В. Лиофилизованный порошок, подходящий для получения раствора для инъекций, получали следующим способом. Общая методика. Наполнители и/или буферирующие агенты, если они используются, растворяют примерно в 60% воды для инъекций. Затем добавляют активное соединение и растворяют, после чего рН доводят до 3-4,5 добавлением 1 М гидроксида натрия и объем доводят до 95% от конечного объема путем добавления воды для инъекций. Если это необходимо, то рН контролируют и корректируют, и объем доводят до конечного объема добавлением воды для инъекций. Затем полученную композицию фильтруют в стерильных условиях через 0,22-микронный фильтр и помещают в стерильный сосуд в условиях асептики. Затем композицию лиофилизуют с использованием соответствующего цикла лиофилизации. Сосуд закрывают крышкой (необязательно, в неполном вакууме или в сухом азоте), помечают и хранят в рефрижераторе. Пример приготовления композиции С. Раствор для инъекций, подходящий для внутривенного введения пациенту, получали из вышеописанной композиции примера В следующим способом. Общая методика. Лиофилизованный порошок композиции примера В (например, содержащий 101000 мг активного соединения) растворяли в 20 мл стерильной воды, после чего полученный раствор разбавляли 80 мл стерильного физиологического раствора в 100-мл контейнере для вливаний. Затем разбавленный раствор внутривенно вводили пациенту в течение 30-120 мин. Применение Перекрестно-сшитые соединения гликопептида-цефалоспорина по настоящему изобретению могут быть использованы в качестве антибиотиков. Так, например, соединения по настоящему изобретению могут быть использованы для лечения или предупреждения бактериальных инфекций и других ассоциированных с бактериями клинических состояний у млекопитающих, включая человека и животныхкомпаньонов (то есть собак, кошек и т.п.), где указанные инфекции и клинические состояния вызываются микроорганизмами, восприимчивыми к соединениям по настоящему изобретению. В соответствии с этим, в одном из своих аспектов, настоящее изобретение относится к способу лечения бактериальной инфекции у млекопитающих, где указанный способ предусматривает введение млекопитающему, нуждающемуся в таком лечении, фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли. Так, например, соединения по настоящему изобретению являются особенно подходящими для лечения или предупреждения инфекций, вызываемых грам-положительными бактериями и родственными микроорганизмами. Например, соединения по настоящему изобретению являются эффективными для лечения или предупреждения инфекций, вызываемых некоторыми бактериями, такими как Enterococcusspp.; Staphylococcus spp., включая коагулазо-отрицательные стафилококки (CNS); Streptococcus spp.; Listeria spp; Clostridium spp; Bacillus spp. и т.п. Примерами видов бактерий, которые могут быть эффективно обработаны соединениями согласно настоящему изобретению, являются, но не ограничиваются ими,резистентные к метициллину бактерии Staphylococcus aureus (MRSA), восприимчивые к метициллину бактерии StapAylococcus aureus (MSSA), восприимчивые к гликопептидному промежуточному соединению бактерии Staphylococcus aureus (GISA), резистентные к метициллину бактерии Staphylococcus epidermitis (MRSE); восприимчивые к метициллину бактерии Staphylococcus epidermitis (MSSE); восприимчивые к ванкомицину бактерии Enterococcus faecalis (EFSVS); восприимчивые к ванкомицину бактерииStreptococcus pyogenes и т.п. Соединения по настоящему изобретению являются менее эффективными,либо вообще неэффективными для лечения или предупреждения инфекций, вызываемых штаммами бактерий, резистентных как к ванкомицину, так и цефалоспорину. Характерными типами инфекций или ассоциированными с бактериальной инфекцией клиническими состояниями, которые могут быть подвергнуты лечению или предупреждению с использованием соединений по настоящему изобретению, являются, но не ограничиваются ими, кожные инфекции или инфекции кожных структур, инфекции мочевых путей, пневмония, эндокардит, трансфузионная инфекция,ассоциированная с введением катетера, остеомиелит и т.п. При лечении таких состояний пациент может- 15007001 быть уже инфицирован микроорганизмом, против которого направлено лечение, либо он просто может быть восприимчивым к инфекции, и в этом случае активный агент вводят в профилактических целях. Соединения по настоящему изобретению обычно вводят в терапевтически эффективном количестве любым подходящим способом введения. Предпочтительно, данные соединения вводят парентерально. Указанные соединения могут быть введены один раз или несколько раз в день. При применении некоторых схем лечения может потребоваться увеличение периодов времени введения, например, такое введение может быть проведено в течение нескольких дней, либо от одной до шести недель или более. Количество активного агента, вводимого за одну дозу, или общее вводимое количество обычно определяет лечащий врач, и это количество зависит от таких факторов, как природа и тяжесть инфекции, возраст и общее здоровье пациента, переносимость пациентом данного активного агента, микроорганизм(ы), вызывающий данную инфекцию, способ введения и т.п. Обычно подходящие дозы составляют в пределах примерно от 0,25 до 10,0 мг/кг/день и предпочтительно примерно от 0,5 до 2 мг/кг/день активного агента. В среднем, для человека весом 70 кг количество активного агента будет составлять примерно от 15 до 700 мг в день или предпочтительно примерно от 35 до 150 мг в день. Кроме того, соединения по настоящему изобретению являются эффективными для ингибирования роста бактерий. В этом варианте осуществления изобретения бактерии подвергают контакту либо in vitro, либо in vivo с ингибирующим рост количеством соединения формулы I или его фармацевтически приемлемой соли. Обычно ингибирующее рост количество будет составлять в пределах примерно от 0,008 до 50 мкг/мл, предпочтительно примерно от 0,008 до 25 мкг/мл и более предпочтительно примерно от 0,008 до 10 мкг/мл. На ингибирование роста бактерий по сравнению с необработанными бактериями обычно указывает снижение или отсутствие размножения бактерий и/или лизис бактерий, то есть снижение колониеобразующих единиц в данном объеме (то есть на мл) в течение данного периода времени (то есть за час) по сравнению с необработанными бактериями. Соединения по настоящему изобретению также являются эффективными для ингибирования биосинтеза клеточной стенки бактерий. В этом варианте осуществления изобретения бактерии подвергают контакту либо in vitro, либо in vivo с определенным количеством соединения формулы I или его фармацевтически приемлемой соли, достаточным для ингибирования биосинтеза клеточной стенки. Обычно количество, ингибирующее биосинтез клеточной стенки, будет составлять в пределах примерно от 0,04 до 50 мкг/мл, предпочтительно примерно от 0,04 до 25 мкг/мл и более предпочтительно примерно от 0,04 до 10 мкг/мл. На ингибирование биосинтеза клеточной стенки бактерий обычно указывает подавление или отсутствие роста бактерий, включая лизис бактерий. Было неожиданно обнаружено, что помимо антибактериальных свойств соединения по настоящему изобретению обладают свойствами, о которых ранее не предполагалось, а именно приемлемой безопасностью для млекопитающих и достаточной растворимостью в воде. Кроме того, было неожиданно обнаружено, что помимо других свойств соединения по настоящему изобретению обладают не предполагаемой ранее быстрой цитолитической активностью против некоторых бактерий, включая резистентные к метициллину бактерии Staphylococci aureus (MRSA) и резистентные к метициллину бактерии Staphylococci epidermitis (MRSE). Указанные свойства, а также ценность соединений по настоящему изобретению как антибиотиков могут быть продемонстрированы с использованием различных in vitro и in vivo анализов, хорошо известных специалистам. Так, например, характерные анализы более подробно описаны в нижеследующих примерах. ПРИМЕРЫ Нижеследующие примеры синтеза и биологических анализов приводятся лишь в целях иллюстрации и не должны рассматриваться как ограничение по настоящему изобретению. В приведенных примерах используются сокращения, которые имеют значения, указанные ниже, если это не оговорено особо. Сокращения, которые не определяются ниже, имеют обычные общепринятые значения. В нижеследующих примерах все температуры выражены в градусах Цельсия (С), если это не оговорено особо. Кроме того, если это не оговорено особо, то все реагенты, исходные материалы и растворители были закуплены у коммерческих поставщиков (таких как Aldrich, Fluka, Sigma и т.п.) и были использованы без дополнительной очистки. Полугидрат гидрохлорида ванкомицина был закуплен у Alpharma, Inc., Fort Lee, NJ 07024 (Alpharma AS, Oslo, Norway). Обращенно-фазовую ВЭЖХ обычно проводили на колонке с C18 и с использованием (А) 98% воды,2% ацетонитрила, 0,1% TFA с избыточным градиентом (например, примерно от 0 до 70%) (В) 10% воды,90% ацетонитрила, 0,1% TFA, если это не оговорено особо. Пример А. Синтез бис-трифторацетатной соли (7R)-7-[(Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(3 аминопропоксиимино)ацетамидо]-3-[(1-пиридинио)метил]-3-цефем-4-карбоксилата. Нижеследующий синтез проиллюстрирован частично в схеме А, представленной выше. Стадия 1. Получение N-(трет-бутоксикарбонил)-3-бромпропиламина (то есть соединения 4, где R1 представляет собой -(СН 2)3-, R2 представляет собой водород, R11 представляет собой ВОС и Z1 представляет собой бром). Гидробромид 3-бромпропиламина (100 г, 457 ммоль) суспендировали в 1,6 л безводного ТГФ. Эту смесь охлаждали до 0 С на бане с ледяной водой и добавляли при интенсивном перемешивании 190 мл триэтиламина. К этой смеси по каплям добавляли трет-бутоксикарбонилангидрид (112,6 г, 516 ммоль) в 200 мл ТГФ. Ледяную баню оставляли для нагревания до комнатной температуры и смесь перемешивали в течение ночи, после чего реакция завершалась, на что указывала ТСХ. Затем смесь фильтровали и фильтрат концентрировали в вакууме. Оставшееся масло разбавляли 1500 мл гексана и хранили при-20 С в течение 3 дней. После этого смесь декантировали и оставшееся твердое вещество сушили в вакууме с получением 101 г (выход 94%) указанного в заголовке промежуточного соединения в виде кристаллического белого твердого вещества. 1H-ЯМР (ДМСО-d6, 300 МГц);1,35-1,39 (с, 9 Н), 1,91-1,95 (м, 2 Н), 2,99-3,04 (т, 2 Н), 3,43-3,52 (т,2 Н), 6,95-6,99 (т, 1 Н). Стадия 2. Получение этил-(Z)-2-(2-трифенилметиламинотиазол-4-ил-2-(3-N-BOC-аминопропоксиимино)ацетата (то есть этилового сложного эфира соединения 5 а, где R1 представляет собой -(СН 2)3-, R2 представляет собой водород, R9 представляет собой трифенилметил, R11 представляет собой ВОС и А представляет собой водород). Гидрохлорид этил-(Z)-2-(2-трифенилметиламино)тиазол-4-ил)-2-(гидроксиимино)ацетата (100 г,202,4 ммоль) растворяли в 700 мл безводного ДМФ. К этой перемешиваемой смеси добавляли карбонат цезия (230,8 г, 708,5 ммоль), а затем йодид тетрабутиламмония (18,7 г, 50,6 ммоль). После этого в течение 30 мин по каплям добавляли N-BOC-3-бромпропиламин (50,6 г, 212,5 ммоль) в ДМФ (100 мл). Смесь перемешивали в течение 2 ч, после чего реакция была завершена, на что указывала ВЭЖХ. После этого смесь фильтровали и осадок на фильтре промывали 200 мл ДМФ. Фильтрат растворяли в 2 л этилацетата и промывали 700 мл 1 н НСl, а затем 700 мл насыщенного водного бикарбоната натрия и, наконец, 500 мл насыщенного раствора соли. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаточное масло растворяли в 250 мл кипящего этанола и выливали в химический стакан. После завершения охлаждения продукта остаточное глинообразное твердое вещество помещали в воронку Бюхнера и промывали 50 мл этанола, предварительно охлажденного до -20 С (Внимание: указанный продукт обладает умеренной растворимостью в этаноле, и его использование в больших количествах будет снижать общий выход конечного продукта). После сушки воздухом остаточное твердое вещество измельчали в ступке с пестиком до получения тонко дисперсного порошка и сушили в вакууме с получением 117 г (выход 94%) указанного в заголовке промежуточного соединения в виде тонкодисперсного не совсем белого порошка.MS m/z: 615,4 [М+Н]+. Стадия 3. Получение (Z)-2-(2-трифенилметиламинотиазол-4-ил)-2-(3-N-ВОС-аминопропоксиимино)ацетата (то есть соединения 5 а, где R1 представляет собой (СН 2)3, R2 представляет собой водород, R9 представляет собой трифенилметил, R11 представляет собой ВОС и А представляет собой водород). Этиловый сложный эфир, полученный в вышеописанной стадии 2 (84,2 г, 137 ммоль), суспендировали в 400 мл безводного этанола и нагревали, перемешивая, в масляной бане до 80 С. После растворения всего материала к раствору в течение 20 мин по каплям добавляли гидроксид калия (23,1 г, 411 ммоль) в 150 мл этанола. Через 10 мин после завершения добавления основания начинал образовываться осадок, а еще через 10 мин смесь становилась твердой. Эту смесь удаляли из масляной бани и охлаждали в ледяной бане. К охлажденной смеси добавляли этилацетат и воду, а затем ее выливали в делительную воронку. Смесь промывали 1 н фосфорной кислотой, что приводило к образованию белого твердого вещества. (Внимание: промывка этого продукта более сильной кислотой, такой как 1 н НСl, будет приводить к разложению продукта). В делительную воронку добавляли воду для растворения этого твердого вещества, а затем органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением указанного в заголовке промежуточного соединения (80 г, выход 99%) в виде темно-коричневого твердого вещества. Стадия 4. Получение (Z)-2-(2-трифенилметиламино-5-хлортиазол-4-ил)-2-(3-N-ВОС-аминопропоксиимино)ацетата (то есть, соединения 5b, где R1 представляет собой -(СН 2)3-, R2 представляет собой водород, R9 представляет собой трифенилметил, R11 представляет собой ВОС и А представляет собой хлор). Промежуточное соединение, полученное на вышеописанной стадии 3 (10 г, 17,04 ммоль), растворяли в 70 мл хлороформа и добавляли при перемешивании твердый N-хлорсукцинимид (2,28 г, 17,04 ммоль). (Внимание: в этих экспериментах предполагается, что избыток NCS может приводить к образованию нежелательных побочных продуктов). Смесь перемешивали в течение ночи (минимум 15 ч), после чего реакция была завершена, на что указывала ВЭЖХ. Затем смесь концентрировали в вакууме и остаток растворяли в минимальном количестве ДМФ. Эту смесь добавляли в интенсивно перемешиваемую воду, в результате чего образовывался осадок, который затем собирали фильтрацией. Твердое вещество сушили воздухом с получением 9,5 г (выход 90%) указанного в заголовке промежуточного соединения в виде коричневого твердого вещества. 1H-ЯМР указывал на присутствие лишь минимального количества остаточного сукцинимида. (Внимание: для успешного проведения реакции сочетания в следующей стадии выделять хлорированный продукт необязательно и в этих экспериментах предполагается, что остаточный сукцинимид может препятствовать последующему замещению пиридина). Альтернативно, после завершения реакции хлорирования реакционную смесь промывали водой (3 х), насыщенным раствором соли, а затем сушили над безводным сульфатом натрия. После этого раствор фильтровали и концентрировали в вакууме с получением указанного в заголовке промежуточного соединения (90%) в виде коричневого твердого вещества. 1(то есть соединения 7, где R1 представляет собой -(СН 2)3-, R2 представляет собой водород, R9 представляет собой трифенилметил, R11 представляет собой ВОС и R12 представляет собой п-метоксибензил). Промежуточное соединение, полученное в стадии 4 (0,62 г, 1 ммоль) растворяли в 6 мл безводного ТГФ и к этой смеси добавляли 0,34 г (0,83 ммоль) гидрохлорида п-метоксибензилового эфира 7-амино-3 хлорметилцефалоспорановой кислоты (то есть соединения 6, где R12 представляет собой РМВ; поставляется Otsuka, Japan) в 4 мл безводного ТГФ. Полученную смесь перемешивали в атмосфере азота и охлаждали до -35 С. К этой охлажденной смеси добавляли диизопропилэтиламин (0,52 мл, 3 ммоль), а затем оксихлорид фосфора (0,11 мл, 1,2 ммоль). Полученную смесь перемешивали при -20 С в течение 30 мин,а затем реакцию гасили сырым ТГФ и разбавляли этилацетатом. После этого смесь промывали водой, 1 н НСl, насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали с получением 0,88 г (выход 100%) указанного в заголовке промежуточного соединения в виде коричневокрасного твердого вещества. 1H-ЯМР не указывал на какую-либо нежелательную изомеризацию и на присутствие остаточного сукцинимида. 1 ЯМP (ДМСО-d6, 300 МГц):1,37 (с, 9 Н), 1,63-1,74 (т, 2 Н), 2,94-2,99 (м, 2 Н), 3,4-3,74 (кв., 2 Н) , 3,75(Внимание: в этих экспериментах предполагается, что при осуществлении вышеуказанной реакции в более крупных масштабах DIPEA вызывает изомеризацию. Эта проблема была решена путем применения модифицированного способа, где в качестве основания использовали 2,4,6-коллидин и где в течение всей реакции, примерно 10 мин, поддерживалась температура -35 С). Стадия 6. Получение п-метоксибензилового эфира (7R)-7-[(Z)-2-(2-трифенилметиламино-5-хлортиазол-4-ил)-2-(3-N-BOC-аминопропоксиимино)ацетамидо]-3-[(1-пиридинио)метил]-3-цефем-4-карбоксилата (то есть соединения 8, где R1 представляет собой -(СН 2)3; R2 представляет собой водород, R9 представляет собой трифенилметил, R11 представляет собой ВОС, R12 представляет собой п-метоксибензил иm равно 0). Промежуточное соединение, полученное в стадии 5 (500 мг, 0,514 ммоль), растворяли в 2 мл безводного ацетона и покрывали фольгой для защиты от света. Раствор перемешивали в атмосфере азота,добавляли 77 мг (0,514 ммоль) йодида натрия и полученную смесь перемешивали в течение 1 ч. Затем добавляли пиридин (63 мкл, 0,772 ммоль) и через 90 мин эту смесь добавляли к 25 мл этилового эфира. Смесь центрифугировали и полученный осадок промывали этиловым эфиром и снова центрифугировали. Эфир декантировали и осадок сушили в вакууме с получением указанного в заголовке промежуточного соединения с количественным выходом в виде коричневого твердого вещества, которое использовали без дополнительной очистки. 1 Н-ЯМР (ДМСО-d6, 300 МГц):1,37 (с, 9 Н), 1,63-1,74 (т, 2 Н), 2,94-2,99 (м, 2 Н), 3,3-3,50 (кв., 2 Н),3,4-3,74 (кв., 2 Н), 3,75 (с, 3H), 3,97-4,05 (т, 2 Н), 5,10-5,12 (д, 1 Н), 5,21 (с, 2 Н), 5,50-5,55 (м, 1 Н), 5,6 (с,2 Н), 6,75-6,81 (т, 1 Н), 6,90-6,96 (д, 2 Н), 7,18-7,41 (м, 17 Н), 8,16-8,21 (т, 2 Н), 8,61-8,70 (т, 1 Н), 8,96 (с, 1 Н),8,98-9,02 (д, 2 Н), 9,41-9,44 (д, 1 Н).MS m/2 1014,2 [М+Н]+. Стадия 7. Получение бис-трифторацетатной соли (7R)-7-[(Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(3 аминопропоксиимино)ацетамидо]-3-[(1-пиридинио)метил]-3-цефем-4-карбоксилата (то есть соединения 2, где R1 представляет собой -(СН 2)3-, R2 представляет собой водород и m равно 0). Промежуточное соединение, полученное в стадии 6 (14,4 г), растворяли в смеси 1:1 трифторуксусной кислоты и дихлорметана (120 мл). К этой перемешанной смеси добавляли 6,2 мл анизола и полученную смесь перемешивали в течение 3 ч при комнатной температуре. После этого смесь концентрировали и остаток растворяли в этилацетате и экстрагировали водой. Водные слои лиофилизовали и полученный порошок растворяли в воде и очищали обращенно-фазовой препаративной ВЭЖХ. Полученный очищенный водный раствор лиофилизовали с получением 3,3 г (выход 30%) указанного в заголовке промежуточного соединения. 1(Внимание: Вышеописанная реакция может быть также проведена с использованием триэтилсилана вместо анизола. Кроме того, указанный продукт может быть выделен путем растирания с этиловым эфиром). Пример В. Синтез бис-трифторацетатной соли (7R)-7-[(Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(3 аминопропоксиимино)ацетамидо]-3-[(2,3-циклопентено-1-пиридинио)метил]-3-цефем-4-карбоксилата. Указанное в заголовке промежуточное соединение получали в соответствии с методом, описанным в примере А, где в стадии 6 вместо пиридина использовали 2,3-циклопентенопиридин (полученный отMS m/z 592,5 [М+Н]+. Пример С. Синтез бис-трифторацетатной соли (7R)-7-[(Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(6-аминогексоксиимино)ацетамидо]-3-[(1-пиридинио)метил]-3-цефем-4-карбоксилата. Указанное в заголовке промежуточное соединение получали в соответствии с методом, описанным в примере А, где в стадии 2 вместо N-BOC-3-бромпропиламина использовали N-BOC-6-йодгексиламин- 19007001 Стадия 2. Получение этил-(Z)-2-(2-трифенилметиламинотиазол-4-ил)-2-[2-(2-N-BOC-аминоэтил) этоксиимино]ацетата (то есть этилового сложного эфира соединения 5 а, где R1 представляет собой-(СН 2)2-O-(СН 2)2-, R2 представляет собой водород, R9 представляет собой трифенилметил, R11 представляет собой ВОС и А представляет собой водород). Промежуточное соединение, полученное в стадии 1 примера А (42,5 г, 86 ммоль), добавляли к перемешиваемой суспензии N-BOC-2-(2-йодэтокси)этиламина (28,5 г, 90 ммоль) (полученной в три стадии из 2-(2-гидроксиэтокси)этанола, то есть (i) BOC2O, KОН, (ii) MsCl, Et3N и (iii) NaI) и карбоната цезия(84,1 г, 258 ммоль) в ДМФ (300 мл). Эту суспензию перемешивали в течение 16 ч при комнатной температуре, после чего реакция завершалась, на что указывала ВЭЖХ. Затем реакционную смесь фильтровали, и осадок на фильтре промывали ДМФ (100 мл). Фильтрат разбавляли этилацетатом (1 л) и промывали водой (300 мл), 1 н НСl (200 мл), насыщенным водным бикарбонатом натрия (200 мл) и насыщенным раствором соли (200 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали колоночной флеш-хроматографией (этилацетат:гексан, 1:1) с получением 49,7 г (выход 90%) указанного в заголовке промежуточного соединения в виде не совсем белого твердого вещества. 1MS m/z 503,1 [М-пиридин]+. Пример Е. Синтез бис-трифторацетатной соли (7R)-7-[(Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(4 аминометилбензилоксиимино)ацетамидо]-3-[(1-пиридинио)метил]-3-цефем-4-карбоксилата. Указанное в заголовке промежуточное соединение получали в соответствии с методом, описанным в примере А и в стадии 2 примера D, где в стадии 2 вместо гидробромида N-BOC-2-(2 йодэтокси)этиламин-3-бромпропиламина использовали N-BOC-4-(йодметил)бензиламин (полученный в четыре стадии из 4-(аминометил)бензойной кислоты, то есть, (i) BOC2O, KОН, (ii) LiAlH4, (iii) MsCl, Et3N и (iv) NaI). 1MS m/z 614,1 [M+H]+, 535,1 [М-пиридин]+. Пример F (сравнительный). Синтез бис-трифторацетатной соли 7(R)-7-[(Z)-2-(2-аминотиазол-4-ил)-2-(3-аминопропоксиимино) ацетамидо]-3-[(1-пиридинио)метил]-3-цефем-4-карбоксилата. Дес-хлоропроизводное промежуточного соединения примера А получали, как описано выше в примере А, но без проведения стадии 4. 1N-(трет-бутоксикарбонил)-3-бромпропиламин (полученный как описано выше в стадии 1 примера А) (9,58 г, 40,23 ммоль) и N-гидроксифталимид (6,36 г, 39 ммоль) растворяли в 70 мл безводного ДМФ. К этому раствору добавляли диизопропилэтиламин (7,01 мл, 40,23 ммоль), в результате чего наблюдалось развитие темно-красной окраски. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, после чего эту реакционную смесь выливали в 500 мл диэтилового эфира. Полученный белый осадок фильтровали и отбрасывали. Органический раствор промывали 2 х 200 мл насыщенного бикарбоната натрия и 2 х 200 мл воды. Органический раствор сушили над безводным сульфатом магния, фильтровали и концентрировали с получением белого твердого вещества. Затем это твердое вещество растворяли в 50 мл ДХМ и 50 мл TFA. После перемешивания в течение 1 ч раствор выливали в 300 мл диэтилового эфира. Полученный осадок фильтровали, промывали диэтиловым эфиром и сушили в вакууме с получением указанного в заголовке промежуточного соединения в виде соли трифторуксусной кислоты. 1H ЯМР (ДМСО-d6, 300 МГц):1,90 (2 Н, кв.), 2,95 (2 Н, т), 4,18 (2 Н, т), 7,79 (4 Н, с), 7,92 (3H, шир. с). Стадия 2. Получение 3-(фталимидоокси)пропиламида ванкомицина. Гидрохлорид ванкомицина (10,0 г, 6,74 ммоль) и промежуточное соединение, полученное в стадии 1 (2,70 г, 8,09 ммоль), суспендировали в 100 мл безводного ДМФ. Затем добавляли диизопропилэтиламин (4,70 мл, 26,98 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 10 мин. Затем добавляли раствор РуВОР (5,61 г, 10,78 ммоль) и HOAt (1,65 г, 10,78 ммоль) в ДМФ (20 мл),и реакционную смесь перемешивали при комнатной температуре. Через 1 ч реакционную смесь добавляли в диэтиловый эфир (500 мл). Полученный осадок фильтровали, промывали диэтиловым эфиром и сушили в вакууме с получением указанного в заголовке промежуточного соединения в виде не совсем белого твердого вещества.- 20007001 МС, m/z: 1651,8 (М+Н)+. Стадия 3. Получение 3-(аминоокси)пропиламида ванкомицина. Промежуточное соединение, полученное в стадии 2 (11,2 г, 6,74 ммоль), суспендировали в 80 мл безводного ДМФ и добавляли моногидрат гидразина (0,65 мл, 13,48 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4,5 ч, после чего к реакционной смеси добавляли 1 мл трифторуксусной кислоты, а затем добавляли 300 мл диэтилового эфира. После интенсивного перемешивания полученный осадок фильтровали, промывали диэтиловым эфиром и сушили в вакууме. Указанное в заголовке соединение очищали обращенно-фазовой ВЭЖХ с использованием градиента вода/метанол,в результате чего получали указанное в заголовке промежуточное соединение в виде лиофилизованного порошка. МС, m/z: 1522,9 (М+Н). Пример Н. Синтез бис-трифторацетата (7R)-7-[2-(2-амино-5-хлортиазол-4-ил)-2-оксоацетамидо]-3-(1-пиридинио)метил-3-цефем-4-карбоксилата. Стадия 1. Получение этил-2-(2-формиламино-5-хлортиазол-4-ил)-2-оксоацетата. Этил-2-(формиламинотиазол-4-ил)-2-оксоацетат (9,1 г, 39,87 ммоль) (полученный от Aldrich, Milwaukee, WI) суспендировали в 50 мл безводного ДМФ. Затем добавляли N-хлорсукцинимид (5,6 г, 41,86 ммоль) в виде твердого вещества и суспензию перемешивали при комнатной температуре. Через 18 ч реакционную смесь выливали в 500 мл воды. Полученный белый осадок фильтровали, промывали водой и сушили воздухом с получением указанного в заголовке промежуточного соединения в виде белого твердого вещества. 1(30 мл, 30 ммоль). Полученную суспензию перемешивали при комнатной температуре в течение 2 ч (после чего раствор становился прозрачным) и добавляли 1 М НСl (30 мл, 30 ммоль), а затем 100 мл воды. После интенсивного перемешивания полученный осадок фильтровали, промывали минимальным количеством холодной воды и сушили воздухом с получением указанного в заголовке промежуточного соединения в виде не совсем белого твердого вещества. 1H-ЯMP (ДMCO-d6, 300 МГц):8,5 (с, 1 Н). Стадия 3. Получение п-метоксибензилового эфира (7R)-7-[2-(2-формиламино-5-хлортиазол-4-ил)-2 оксоацетамидо]-3-хлорметил-3-цефем-4-карбоксилата. Промежуточное соединение, полученное в стадии 2 (1,03 г, 4,37 ммоль), гидрохлорид пметоксибензилового эфира 7-амино-3-хлорметилцефалоспорановой кислоты (1,95 г, 4,81 ммоль) и HOAt(0,74 г, 4,81 ммоль) суспендировали в 15 мл безводного ДМФ. Реакционный сосуд продували азотом, а затем охлаждали до 0 С с использованием внешней ледяной бани. К охлажденной реакционной смеси добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,92 г, 4,81 ммоль), а затем добавляли 2,4,6-коллидин (0,64 мл, 4,81 ммоль). Реакционную смесь перемешивали при 0 С в течение 2 ч и затем выливали в 200 мл 0,5 М НСl. Полученный осадок фильтровали, промывали водой и сушили воздухом с получением указанного в заголовке промежуточного соединения в виде красного твердого вещества. Это соединение использовали без дополнительной очистки. МС, m/z: 607 (M+Na)+. Стадия 4. Получение п-метоксибензилового эфира (7R)-7-[2-(2-формиламино-5-хлортиазол-4-ил)-2 оксоацетамидо]-3-(1-пиридинио)метил-3-цефем-4-карбоксилата. Промежуточное соединение, полученное в стадии 3 (2,5 г, 4,27 ммоль), и йодид натрия (0,64 г, 4,27 ммоль) растворяли в ацетоне и покрывали фольгой для защиты от света. Реакционную смесь перемешивали 10 мин и затем добавляли пиридин (0,42 мл, 5,12 ммоль). Затем реакционную смесь перемешивали при комнатной температуре в течение 1 ч, после чего добавляли 300 мл воды. Полученный осадок фильтровали, промывали водой и сушили воздухом с получением красного твердого вещества. Это твердое вещество очищали обращенно-фазовой ВЭЖХ, и полученный водный раствор лиофилизовали с получением указанного в заголовке промежуточного соединения в виде лиофилизованного порошка. МС, m/z: 628,1 (М)+. Стадия 5. Получение бис-трифторацетата (7R)-7-[2-(2-амино-5-хлортиазол-4-ил)-2-оксоацетамидо]3-(1-пиридинио)метил-3-цефем-4-карбоксилата. Промежуточное соединение, полученное в стадии 4 (0,11 г, 0,18 ммоль), растворяли в 5 мл метанола и добавляли концентрированную водную соляную кислоту (0,5 мл). Полученный раствор перемешивали при комнатной температуре в течение 1,5 ч. Метанол удаляли в вакууме и добавляли ацетонитрил(10 мл). Затем раствор концентрировали в вакууме, к остатку добавляли ДХМ (2 мл) и TFA (2 мл) и полученную смесь перемешивали при комнатной температуре в течение 1,5 ч. После этого добавляли диэтиловый эфир (50 мл) и указанное в заголовке промежуточное соединение выделяли центрифугированием. Это промежуточное соединение использовали без дополнительной очистки.- 21007001 МС, m/z: 479,9 (М)+. Пример 1. Синтез соединения 13, где R1 представляет собой -(СН 2)3-, R2, R5, R6, R8 представляют собой водород, R4 представляет собой гидрокси, R7 представляет собой метил, Х 1 и Х 2 представляют собой хлор и m равно 0. Стадия 1. Получение (Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(3-аминопропоксиимино)ацетата. Промежуточное соединение, полученное в стадии 4 примера А (0,75 г, 1,21 ммоль), растворяли в 5 мл ДХМ и 5 мл трифторуксусной кислоты. После перемешивания в течение 1 ч при комнатной температуре добавляли 100 мл диэтилового эфира. Полученный осадок фильтровали, промывали диэтиловым эфиром и сушили в вакууме с получением указанного в заголовке промежуточного соединения в виде коричневого твердого вещества. Стадия 2. Получение соединения 13, где R1 представляет собой -(СН 2)3-, R2, R5, R6, R8 представляют собой водород, R4 представляет собой гидрокси, R7 представляет собой метил, Х 1 и X2 представляют собой хлор и m равно 0. Гидрохлорид ванкомицина (1,3 г, 0,88 ммоль) и HOAt (0,14 г, 0,88 ммоль) суспендировали в 3,5 мл безводного ДМСО. Затем добавляли раствор РуВОР (0,46 г, 0,88 ммоль) в 3,5 мл безводного ДМФ, после чего добавляли DIPEA (154 мкл, 0,88 ммоль). После перемешивания в течение 20 мин добавляли раствор промежуточного соединения, полученного в стадии 1 (0,22 г, 0,44 ммоль), в 1 мл ДМФ, а затем быстро добавляли DIEA (0,54 мл, 3,08 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, после чего добавляли 0,5 мл трифторуксусной кислоты, а затем быстро добавляли 100 млEt2O. Полученный осадок фильтровали, промывали Et2O и сушили в вакууме. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и полученный водный раствор лиофилизовали с получением указанного в заголовке промежуточного соединения в виде лиофилизованного порошка. МС, m/z: 1711,0 (М+Н)+. Пример 1. Синтез соединения формулы I, где R1 представляет собой -(СН 2)3-, R2, R5, R6, R8 представляют собой водород, R4 представляет собой гидрокси, R7 представляет собой метил, X1 и X2 представляют собой хлор и m равно 0 (соединение 1 в табл. 1) Гидрохлорид ванкомицина (4,2 г, 2,8 ммоль) растворяли в 40 мл ДМСО. К полученному раствору добавляли раствор РуВОР (1,3 г, 2,6 ммоль) и HOAT (0,35 г, 2,6 ммоль) в 40 мл ДМФ, а затем добавляли 0,98 мл (5,68 ммоль) диизопропилэтиламина. Смесь перемешивали при комнатной температуре в течение 30 мин, а затем реакцию гасили 0,44 мл (5,7 ммоль) трифторуксусной кислоты. После этого смесь охлаждали до 0 С и добавляли раствор промежуточного соединения, полученного, как описано выше в примере А (1,3 г, 2,6 ммоль), в 20 мл ДМФ при 0 С, а затем добавляли 1,5 мл (11,4 ммоль) 2,4,6-коллидина. Смесь выдерживали при 0 С в течение 4 ч, а затем реакцию гасили 1,1 мл трифторуксусной кислоты. Эту смесь добавляли к этиловому эфиру, в результате чего образовывался осадок, который затем центрифугировали, промывали эфиром, декантировали и сушили в вакууме. Полученный порошок растворяли в воде и очищали препаративной ВЭЖХ. Фракции, содержащие нужный продукт, лиофилизовали, в результате чего получали соль три-трифторуксусной кислоты указанного в заголовке соединения. Анион указанной соли затем подвергали ионному обмену с использованием смолы Амберлит и получали тригидрохлоридную соль указанного в заголовке соединения (1,4 г, выход 27%) в виде белого порошка. МС, m/z: 953,3 М+Н]+-пиридин]/2; 992,0 [М+Н]+/2. Кроме того, соединения 2-30, представленные в табл. 1, были получены в соответствии с методами примера А и примера 1, где вместо пиридина в стадии 6 примера А использовали следующие замещенные пиридины: Пример 2 - 2-пиколин. Пример 3 - 3-пиколин. Пример 4 - 4-пиколин. Пример 5 - 2-метоксипиридин. Пример 6 - 3-метоксипиридин. Пример 7 - 4-метоксипиридин. Пример 8 - 2-тиометоксипиридин. Пример 9 - 3-тиометоксипиридин. Пример 10 - 4-тиометоксипиридин. Пример 11 - 2-фторпиридин. Пример 12 - 3-фторпиридин. Пример 13 - 4-фторпиридин. Пример 14 - 2-хлорпиридин. Пример 15 - 3-хлорпиридин. Пример 16 - 4-хлорпиридин. Пример 17 - 2-фенилпиридин. Пример 18 - 3-фенилпиридин. Пример 19 - 4-фенилпиридин. Пример 20 - 4-циклопропилпиридин.- 22007001 Пример 21 - 4-(карбокситиометокси)пиридин. Пример 22 - изоникотинамид. Пример 23 - 2,3-лутидин. Пример 24 - 3,4-лутидин. Пример 25 - 3,5-лутидин. Пример 26 - 3,4-диметоксипиридин. Пример 27 - 4-метокси-3-метилпиридин. Пример 28 - 4-фтор-3-метоксипиридин. Пример 29 - 2,3-циклогексенопиридин. Пример 30 - 2,3-циклопентенопиридин. Вышеуказанные замещенные пиридины либо являются коммерчески доступными, либо они могут быть получены в соответствии с методами, описанными в литературе. Пример 31. Синтез соединения формулы I, где R1 представляет собой -(СН 2)6-, R2, R5, R6, R8 представляют собой водород, R4 представляет собой гидрокси, R7 представляет собой метил, X1 и X2 представляют собой хлор и m равно 0 (соединение 31 в табл. 1). В соответствии с методом примера 1 и с использованием промежуточного соединения примера С вместо промежуточного соединения примера А получали указанное в заголовке соединение. МС, m/z: 2026,5 (М+)+. Пример 32. Синтез соединения формулы I, где R1 представляет собой -(СН 2)2-O-(СН 2)2-, R2, R5, R6,8R представляют собой водород, R4 представляет собой гидрокси, R7 представляет собой метил, X1 и X2 представляют собой хлор и m равно 0 (соединение 32 в табл. 1). В соответствии с методом примера 1 и с использованием промежуточного соединения примера D вместо промежуточного соединения примера А получали указанное в заголовке соединение. МС, m/z: 967,9 [(М-пиридин)/2)]+. Пример 33. Синтез соединения формулы I, где R1 представляет собой -СН 2-1,4-Рh-СН 2; R2, R5, R6,8R представляют собой водород, R4 представляет собой гидрокси, R7 представляет собой метил, X1 и X2 представляют собой хлор и m равно 0 (соединение 33 в табл. 1). В соответствии с методом примера 1 и с использованием промежуточного соединения примера Е вместо промежуточного соединения примера А получали указанное в заголовке соединение.-(СН 2)3-, R2, R5, R6, R8 представляют собой водород, R4 представляет собой гидрокси, R7 представляет собой метил, Х 1 и X2 представляют собой хлор и m равно 0 (соединение 34). В соответствии с методом примера 1 и с использованием промежуточного соединения дес-хлороцефалоспорина примера F вместо промежуточного соединения примера А получали указанное в заголовке соединение.MC, m/z: 935,3 М+Н]+-пиридин]/2; 974,9 [М+Н]+/2. Пример 35. Определение минимальных ингибирующих концентраций (MIC). Анализы на определение минимальных ингибирующих концентраций (MIC) осуществляли методом микроразведений в бульоне, как описано в руководствах NCCLS (см. NCCLS. 2000. Methods for DilutionAntimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard - Fifth Ed., Vol. 20,No.2). Бактериальные штаммы были получены из Американской коллекции типовых тканевых культур(АТСС), из клиники при Стэнфордском университете (SU), из Кайзеровской постоянной региональной лаборатории в Беркли (КРВ), из Массачусетской больницы общего профиля (MGH), из Центра по контролю за заболеваниями (CDC), из Сан-Францисского административного госпиталя для ветеранов(SFVA) или из Сан-Францисской клиники при Калифорнийском университете (UCSF). Резистентные к ванкомицину энтерококки были фенотипированы как Van А или Van В на основе их чувствительности к тейкопланину. Некоторые резистентные к ванкомицину энтерококки, которые были генотипированы какVan A, Van В, Van C1 или Van C2, были также получены из клиники Майо. В этом анализе криоконсервированные бактериальные культуры эталонных и клинических штаммов высевали штрихом на соответствующую агаровую среду для выделения (то есть на триптиказосоевый агар, на триптиказо-соевый агар с дефибринированными бараньими эритроцитами, на агар с сердечно-мозговым экстрактом, на шоколадный агар). После инкубирования для образования колоний эти планшеты герметично закрывали парафильмом и хранили в холодильнике в течение до двух недель. В целях приготовления инокулята для анализа и в целях обеспечения низкой вариабельности несколько колоний от бактериального изолята, культивируемого на планшетах с агаром, забирали инокулирующей петлей и асептически переносили на бульон Меллера-Хинтона (в который добавляли двухвалентные катионы до требуемых уровней в соответствии с инструкцией производителя). Бульонную культуру выращивали в течение ночи при 35 С, разводили в свежем предварительно нагретом бульоне и культивировали до лог-фазы, которая была эквивалентна 0,5 стандарту MacFarland или 1 х 108 колониеобразующим единицам на один миллилитр (к.о.е./мл). Из-за вариабельности видов не все клеточные суспензии содержали 1 х 108 к.о.е./мл, даже если мутность клеток была эквивалентна стандарту MacFarland, и поэтому в- 23007001 разведениях различных бактериальных штаммов была сделана приемлемая коррекция (в соответствии с руководством NCCLS). Инокулят разводили так, чтобы 100 мкл этой культуры в бульоне МеллераХинтона, дополненном бульоне Меллера-Хинтона или в тест-среде на Haemophilus, при нанесении на 2 кратные серийные разведения концентраций антибиотика, приготовленные также в 100 мкл соответствующей среды в 96-луночном микротитрационном планшете, давали исходную концентрацию бактерий 5 х 105 к.о.е./мл. Затем эти планшеты инкубировали в течение 18-24 ч при 35 С. MIC означает определяемую визуально самую низкую концентрацию, которая полностью подавляет рост бактерий. На рост бактерий указывает наличие более чем трех небольших колоний, наличие скопления осажденных клеток более чем 2 мм в диаметре, или визуально наблюдаемая мутность клеточной культуры. Штаммами, тестируемыми в начальном скрининге рутинным способом, были чувствительные к метициллину бактерии Staphylococcus aureus (MSSA); резистентные к метициллину бактерии Staphylococcus aureus (MRSA); бактерии Staphylococcus aureus, продуцирующие пенициллиназу; чувствительные к метициллину бактерии Staphylococcus epidermidis (MSSE); резистентные к метициллину бактерии(EFMVS); чувствительные к ванкомицину бактерии Enterococcus faecalis (EFSVS); резистентные к ванкомицину бактерии Enterococcus faecium, которые также являются резистентными к тейкопланину(EFMVR Van A); резистентные к ванкомицину бактерии Enterococcus faecium, которые являются чувствительными к тейкопланину (EFSMR Van В); резистентные к ванкомицину бактерии Enterococcusfaecalis, которые также являются резистентными к тейкопланину (EFSVR Van A); резистентные к ванкомицину бактерии Enterococcus faecalis, которые являются чувствительными к тейкопланину (EFSVR Van В); чувствительные к пенициллину бактерии Streptococcus pneumoniae (PSSP) и резистентные к пенициллину бактерии Streptococcus pneumoniae (PSRP). Поскольку PSSP и PSRP не способны к хорошему росту на бульоне Меллера-Хинтона, то MIC для этих штаммов определяли с использованием либо бульона TS,в который добавляли дефибринированную кровь, либо тест-среды для Haemophilus. Затем тестируемые соединения, обладающие значительной активностью против вышеупомянутых штаммов, анализировали в целях определения значений MIC для более широкой панели клинических изолятов, включая виды, перечисленные выше, а также невидовые коагулазо-отрицательные Staphylococcus, как резистентные, так и чувствительные к метициллину (MS-CNS и MR-CNS). Кроме того, эти тестируемые соединения были также проанализированы на MIC против грам-отрицательных микроорганизмов, таких как Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae, Enterobacter cloacae,Acinetobacter baumannii, Haemophilus influenzae и Moraxella catarrhalis. В табл. 2 представлены данные МIC90 для соединения по настоящему изобретению, обладающего активностью против резистентных к метициллину бактерий S. aureus (MRSA) и резистентных к метициллину бактерий S. epidermitis (MRSE), по сравнению с известным антибиотиком ванкомицином. Таблица 2. Минимальные ингибирующие концентрации (MIC) Число протестированных штаммов Минимальная ингибирующая концентрация для 90% протестированных штаммов Кроме того, как показано в табл. 3, соединения по настоящему изобретению по сравнению с родственным дес-хлоропроизводным (то есть соединением 34) также неожиданно обнаруживали ранее не предполагаемые MIC против различных резистентных к метициллину штаммов S.aureus. Пример 36. Анализ зависимости уничтожения бактерий от времени. Анализ зависимости уничтожения бактерий от времени представляет собой метод определения скорости бактерицидного действия тестируемого соединения. Эти методы аналогичны методам, описаннымV. Lorian, "Antibiotics in Laboratory Medicine", Fourth Edition, WilliamsWilkins (1996), pages 104-105. Для скорейшего предупреждения бактериальной колонизации и снижения степени заражения ткани хозяина желательно быстрое уничтожение бактерий. Бактериальные инокуляты получали, как описано в примере 35, для определения MIC. Бактерии разводили в предварительно нагретых средах в шейкерных колбах и инкубировали при встряхивании(200 об/мин, 35 С). Через 0, 1, 4 и 24 ч из колб брали пробы и бактерии подсчитывали на планшетесчетчике. После первоначального забора проб к культурам в шейкерных колбах добавляли тестируемое соединение. Число бактерий в планшетах до и после добавления соединения выражали графически в виде кривой зависимости уничтожения бактерий от времени. Считалось, что соединение имеет бактерицидную активность, если log снижения числа бактериальных клеток за 24 ч составляет 3 (снижение,равное 99,9% или более). В этом анализе соединение формулы I, то есть соединение 1, имело бактерицидное действие противMSSA 13709 и MRSA 33591 при концентрации 1 мкг/мл за 4 ч. В отличие от этого, ванкомицин оказывал бактерицидное действие против MSSA 13709 и MRSA 33591 при концентрации 4 мкг/мл за 24 ч. Пример 37. Исследования in vivo-эффективности у мышей с нейтропенией. Животных (самцов мышей CD-1, 20-30 г) приобретали в лаборатории Чарльза Ривера (Gilroy, CA) и давали корм и воду ad libitum. Нейтропению индуцировали путем внутрибрюшинной (i.p.) инъекции 200 мг/кг циклофосфамида за четыре и за два дня до инокуляции бактериями. Используемыми микроорганизмами были либо чувствительные, либо резистентные штаммы клинически релевантных грам-положительных патогенов, такие как чувствительные к метициллину бактерии Staphylococcus aureus (MSSA 13709) и резистентные к метициллину бактерии Staphylococcus aureus(MRSA 33591). Концентрация бактериального инокулята составляла 106 к.о.е./мл. Животных подвергали легкой анестезии изофлураном и в переднюю область бедра инъецировали 50 мл указанного бактериального инокулята. Через один час после инокуляции животным внутривенно вводили дозу носителя или соответствующую дозу тестируемого соединения. На час 0 и через 24 ч после обработки животных подвергали эвтаназии (путем СO2-асфиксии) и вынимали переднюю и заднюю части бедра в условиях асептики. Это бедро помещали в 10 мл стерильного физиологического раствора и гомогенизировали. Разведения гомогената высевали на планшеты с триптиказо-соевым агаром, которые инкубировали в течение ночи. Число бактериальных колоний в данном планшете умножали на число разведений, делили на массу бедра (в граммах) и результат выражали как log к.о.е./г. Для каждого тестируемого соединения вычисляли ED50 (дозу, необходимую для продуцирования 50% от максимального снижения титра в бедре). В этом анализе с использованием MRSA 33591 соединение формулы I, т.е. соединение I имелоED500,20 мг/кг, i.v., тогда как для ванкомицина ED50 составляла 9 мг/кг, i.v. Пример 38. Определение растворимости в воде. Растворимость в воде соединения по настоящему изобретению определяли в соответствии с нижеследующим методом. 5 мас.% декстрозного буферного раствора при рН 2,2 получали путем добавления 1 мл 1 н соляной кислоты (Aldrich) к 99 мл 5 мас.% водного раствора декстрозы (Baxter). Затем для получения калибровочных стандартов приготавливали 1 мг/мл маточного раствора путем растворения 1 мг тестируемого соединения в 1 мл ДМСО. Этот раствор интенсивно перемешивали в течение 30 с, а затем обрабатывали ультразвуком в течение 10 мин. После этого маточный раствор разводили водой для получения калибровочных стандартов, имеющих следующие концентрации: 50, 125, 250,375 и 500 мкг/мл.- 25007001 Каждое тестируемое соединение (30 мг) загружали в миллипоровый нестерильный 0,1 мкм фильтрUltrafree-MC (Millipore UFC30VVOO) и каждый фильтр снабжали магнитной мешалкой. Затем в каждый фильтр добавляли 5 мас.% декстрозного буферного раствора (750 мкл) и полученные смеси интенсивно перемешивали в течение 5 мин. После этого фильтры помещали в пробирки Эппендорфа на подставке и эту подставку с пробирками помещали в верхнюю часть магнитной мешалки. Затем каждый фильтр титровали до рН 3 с использованием 1 н NaOH (VWR) и полученные растворы центрифугировали при 7000 об/мин в течение 5 мин. После этого каждый фильтр 200-кратно разводили 5% декстрозным буферным раствором и разведенные образцы переносили в автоматический аппликатор образцов для анализа. Калибровочные стандарты и тестируемые образцы анализировали с помощью обращенно-фазовой ВЭЖХ при следующих условиях: Колонка Luna 150 х 4,6 мм; С 18; 5u Подвижная фаза А=5/95, В=95/5, оба=МеСМ/Н 2O; 0,1% TFA Метод 10m Lido 100 (0-100% В за 6 мин) Объем впрыска 20 мкл Длина волны 214 нм Растворимость каждого тестируемого образца вычисляли путем сравнения площадей под пиком данного тестируемого образца для построения калибровочной кривой и умножали на число разведении. С использованием вышеописанного метода для препаратов образцов в дубликатах было обнаружено, что соединение 1 имеет растворимость 47,9 мг/мл. Хотя настоящее изобретение описано выше со ссылками на конкретные варианты его осуществления, однако для каждого специалиста очевидно, что в него могут быть внесены различные эквивалентные изменения, не выходящие за рамки существа и объема настоящего изобретения. Кроме того, может быть внесено множество модификаций для адаптации конкретной ситуации, конкретных материалов,композиций, конкретного способа, стадии или стадий конкретного способа, применительно к целям, объему и существу настоящего изобретения. Все указанные модификации должны быть в пределах объема,определенного в прилагаемой формуле изобретения. Кроме того, все публикации, патенты и патентные документы, цитируемые в настоящей заявке, во всей своей полноте вводятся в настоящее описание посредством ссылки так, как если бы они по отдельности были введены посредством ссылки. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы I или его фармацевтически приемлемая соль,где X1 и X2 независимо выбраны из группы, состоящей из водорода и хлора;Ya и Yb независимо представляют собой C1-6 алкилен;R2, R5, R6 и R8 представляют собой водород; каждый R3 независимо выбран из группы, состоящей из C1-6 алкила, С 2-6 алкенила, С 2-6 алкинила, С 3-6 циклоалкила, С 6-10 арила, С 2-9 гетероарила, С 3-6 гетероциклила и Ra, либо две смежные группы R3, взятые вместе, образуют С 3-6 алкилен или -О-(C1-6 алкилен)-О-, где каждая алкильная, алкиленовая, алкенильная и алкинильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы,- 26007001 состоящей из Ra и Rc; и каждая арильная, циклоалкильная, гетероарильная и гетероциклическая группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Rb; где каждый Ra независимо выбран из группы, состоящей из -ORd, галогена, -SRd, -S(O)Rd, -S(O)2Rd,-S(О)2ORd, -S(О)2NRdRe, -NRdRe, -CO2Rd, -ОС(O)Rd, -C(O)NRdRe, -NRdC(O)Re, -OC(O)NRdRe,-NRdC(O)ORe, -NRdC(O)NRdRe, -СF3 и -ОСF3; каждый Rb независимо выбран из группы, состоящей из C1-6 алкила, С 2-6 алкенила, С 2-6 алкинила и Ra; каждый Rc независимо выбран из группы, состоящей из С 3-6 циклоалкила, С 6-10 арила, С 2-9 гетероарила и С 3-6 гетероциклила, где каждая циклоалкильная, арильная, гетероарильная и гетероциклильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из C1-6 алкила и Rf; каждый Rd и Re независимо выбран из группы, состоящей из водорода, C1-6 алкила, С 2-6 алкенила, С 2-6 алкинила, С 3-6 циклоалкила, С 6-10 арила, С 12-9 гетероарила и С 3-6 гетероциклила, либо Rd и Re, взятые вместе с атомами, с которыми они связаны, образуют С 3-6 гетероциклическое кольцо, имеющее 1-3 гетероатома,независимо выбранных из кислорода, азота или серы, где каждая алкильная, алкенильная и алкинильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Rc и Rf, и каждая арильная, циклоалкильная, гетероарильная и гетероциклильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из C1-6 алкила и Rf; каждый Rf независимо выбран из группы, состоящей из -ОН, -OC1-6 алкила, -SC1-6 алкила, -F, -Cl,-NH2, -NН(С 1-6 алкила), -N(C1-6 алкила)2, -ОС(О)C1-6 алкила, -С(O)ОС 1-6 алкила, -NHC(О)C1-6 алкила,-С(О)ОН, -С(О)NH2, -С(O)NHC1-6 алкила, -С(О)N(C1-6 алкила)2, -СF3 и -ОСF3; где указанные гетероарильная и гетероциклильная группы содержат от 1 до 3 гетероатомов, выбранных из азота, кислорода или серы;n равно 0 или 1. 2. Соединение по п.1, где n равно 0. 3. Соединение по п.1, где n равно 0 и Ya и Yb, взятые вместе, образуют группу -(СН 2)2-8-. 4. Соединение по п.3, где n равно 0 и Ya и Yb, взятые вместе, образуют группы -(СН 2)2-, -(СН 2)3-,-(СН 2)4-, -(СН 2)5- или -(СН 2)6-. 5. Соединение по п.4, где n равно 0 и Ya и Yb, взятые вместе, образуют группу -(СН 2)3-. 6. Соединение по п.1, где n равно 1. 7. Соединение по п.1, где n равно 1 и Ya и Yb оба представляют собой -СН 2-. 8. Соединение по п.7, где W представляет собой фенилен. 9. Соединение по п.1, где n равно 1, Ya и Yb оба представляют собой -СН 2 СН 2- и W представляет собой -О-. 10. Соединение по любому из пп.1-9, где m равно 0. 11. Соединение по любому из пп.1-9, где m равно 1 или 2 и каждый R3 независимо выбран из группы, состоящей из C1-6 алкила, С 3-6 циклоалкила, -ORd, -SRd, -F или -Сl; либо две смежные группы R3, взятые вместе, образуют С 3-6 алкилен. 12. Соединение по любому из пп.1-11, где оба X1 и X2 представляют собой хлор. 13. Соединение по п.1, где R1 представляет собой -Ya-(W)n-Yb-, где n равно 0 и Ya и Yb, взятые вместе, образуют группу -(СН 2)3-, оба X1 и X2 представляют собой хлор и m равно 0. 14. Соединение по п.1, где оба X1 и X2 представляют собой хлор и R1, R2, R3 и m такие, как определено в табл. 1. 15. Соединение формулы II или его соль,где Р 1 и Р 2 независимо представляют собой водород или аминозащитную группу; Р 3 представляет собой водород или карбоксизащитную группу;Q представляет собой удаляемую группу или группу формулыYa и Yb независимо представляют собой C1-6 алкилен;R2 представляет собой водород; каждый R3 независимо выбран из группы, состоящей из C1-6 алкила, С 2-6 алкенила, С 2-6 алкинила, С 3-6 циклоалкила, С 6-10 арила, С 2-9 гетероарила, С 3-6 гетероциклила и Ra, либо две смежные группы R3, взятые вместе, образуют С 3-6 алкилен или -О-(C1-6 алкилен)-О-, где каждая алкильная, алкиленовая, алкенильная и алкинильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы,состоящей из Ra и Rc; и каждая арильная, циклоалкильная, гетероарильная и гетероциклическая группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Rb; каждый Ra независимо выбран из группы, состоящей из -ORd, галогена, -SRd, -S(O)Rd, -S(O)2Rd,-S(O)2ORd, -S(O)2NRdRe, -NRdRe, -CO2Rd, -OC(O)Rd, -C(O)NRdRe, -NRdC(O)Re, -OC(O)NRdRe,-NRdC(O)ORe, -NRdC(O)NRdRe, СF3 и -ОСF3; каждый Rb независимо выбран из группы, состоящей из C1-6 алкила, С 2-6 алкенила, С 2-6 алкинила и Ra; каждый Rc независимо выбран из группы, состоящей из С 3-6 циклоалкила, С 6-10 арила, С 2-9 гетероарила и С 3-6 гетероциклила, где каждая циклоалкильная, арильная, гетероарильная и гетероциклильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из C1-6 алкила и Rf; каждый Rd и Re независимо выбран из группы, состоящей из водорода, C1-6 алкила, С 2-6 алкенила, С 2-6 алкинила, С 3-6 циклоалкила, С 6-10 арила, С 2-9 гетероарила и С 3-6 гетероциклила, либо Rd и Re, взятые вместе с атомами, с которыми они связаны, образуют С 3-6 гетероциклическое кольцо, имеющее 1-3 гетероатома,независимо выбранных из кислорода, азота или серы, где каждая алкильная, алкенильная и алкинильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из Rc и Rf, и каждая арильная, циклоалкильная, гетероарильная и гетероциклильная группа необязательно замещена 1-3 заместителями, независимо выбранными из группы, состоящей из C1-6 алкила и Rf; каждый Rf независимо выбран из группы, состоящей из -ОН, -OC16 алкила, -SC1-6 алкила, -F, -Cl,-NH2, -NH(C1-6 алкила), -N(С 1-6 алкила)2, -ОС(О)C1-6 алкила, -С(О)OC1-6 алкила, -NHC(O)С 1-6 алкила,-С(О)ОН, -C(O)NH2, -С(О)NHC1-6 алкила, -С(О)N(C1-6 алкила)2, -СF3 и -ОСF3; Х- представляет собой необязательно присутствующий анион. или его соли,где Р 1 и Р 2 независимо представляют собой водород или аминозащитную группу; Р 4 представляет собой водород или карбоксизащитную группу;Ya и Yb независимо представляют собой C1-6 алкилен;R2 представляет собой водород иn равно 0 или 1. 17. Фармацевтическая композиция для лечения бактериальной инфекции у млекопитающего, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по любому из пп.1-14. 18. Способ лечения бактериальной инфекции у млекопитающего, где указанный способ предусматривает введение указанному млекопитающему фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по любому из пп.114. 19. Способ ингибирования роста бактерий, где указанный способ предусматривает контактирование бактерий с ингибирующим рост количеством соединения по любому из пп.1-14. 20. Способ ингибирования биосинтеза клеточной стенки бактерий, где указанный способ предусматривает контактирование бактерий с определенным количеством соединения по любому из пп.1-14,ингибирующим биосинтез клеточной стенки. 21. Способ получения соединения по любому из пп.1-14, где указанный способ предусматривает взаимодействие гликопептида формулы 1 или его соли с соединением формулы 2 или его солью с получением соединения формулы I или его соли. 22. Способ получения соединения по любому из пп.1-14, где указанный способ предусматривает взаимодействие соединения формулы 10 или его соли с соединением формулы 11 или его солью с получением соединения формулы I или его соли. 23. Способ получения соединения по любому из пп.1-14, где указанный способ предусматривает взаимодействие соединения формулы 9 или его соли с соединением формулы 13 или его солью с получением соединения формулы I или его соли. 24. Продукт, полученный способом по любому из пп.21-23. 25. Применение соединения по любому из пп.1-14, при приготовлении лекарственного средства для лечения бактериальной инфекции у млекопитающего.
МПК / Метки
МПК: C07D 501/00, C07K 9/00, A61P 31/00, A61K 31/545, C07D 501/20, A61K 38/04
Метки: антибиотики, гликопептид-цефалоспорин, перекрестно-сшитые
Код ссылки
<a href="https://eas.patents.su/30-7001-perekrestno-sshitye-antibiotiki-glikopeptid-cefalosporin.html" rel="bookmark" title="База патентов Евразийского Союза">Перекрестно-сшитые антибиотики “гликопептид-цефалоспорин”</a>
Предыдущий патент: Способ получения периндоприла, его аналогов и солей с использованием 2,5-диоксо-оксазолидиновых промежуточных соединений
Следующий патент: Одностадийный способ получения 3,4,5-триметокситолуола
Случайный патент: Лечение ревматоидного заболевания глюкокортикоидами с отсроченным высвобождением