Устойчивые оральные фармацевтические дозированные формы, содержащие опиоидный анальгетик
Номер патента: 18311
Опубликовано: 30.07.2013
Авторы: О'доннэлл Эдвард Патрик, Мэннион Ричард Оуэн, Хуан Хайюн Хью, Маккенна Уильям Генри





























Формула / Реферат
1. Способ изготовления твердой лекарственной формы для перорального применения с пролонгированным высвобождением, включающий, по меньшей мере, следующие стадии:
(a) объединение (1) по меньшей мере одного полиэтиленоксида, имеющего, по данным реологических измерений, молекулярную массу по меньшей мере 1000000, и (2) по меньшей мере одного активного ингредиента с образованием композиции;
(b) формование композиции с образованием матричной формы лекарственного препарата с пролонгированным высвобождением;
(c) отверждение указанной матричной формы лекарственного препарата с пролонгированным высвобождением, включающее, по меньшей мере, стадию отверждения, на которой матричную форму препарата с пролонгированным высвобождением нагревают до температуры, которая является, по меньшей мере, температурой размягчения указанного полиэтиленоксида, в течение по меньшей мере 1 мин.
2. Способ по п.1, в котором на стадии (c) матричную форму препарата с пролонгированным высвобождением нагревают до температуры, которая является, по меньшей мере, температурой размягчения указанного полиэтиленоксида, в течение по меньшей мере 5 мин.
3. Способ по п.1 или 2, в котором на стадии (b) композицию формуют с образованием матричной формы лекарственного препарата с пролонгированным высвобождением в виде таблетки.
4. Способ по п.3, в котором на стадии (b) композицию формуют непосредственным прессованием указанной композиции.
5. Способ по любому из пп.1-4, в котором на стадии (c) матричную форму препарата с пролонгированным высвобождением нагревают до температуры по меньшей мере 60 или по меньшей мере 62°С.
6. Способ по п.5, в котором матричную форму препарата с пролонгированным высвобождением нагревают до температуры от 62 до 90°С, от 65 до 90°С или от 68 до 90°С.
7. Способ по п.5, в котором матричную форму препарата с пролонгированным высвобождением нагревают до температуры по меньшей мере 62 или по меньшей мере 68°С в течение от 1 до 5 мин или от 5 мин до 3 ч.
8. Способ по п.6, в котором матричную форму препарата с пролонгированным высвобождением нагревают до температуры по меньшей мере 60°С, или по меньшей мере 62°С, или от 62 до 85°С в течение по меньшей мере 15, или 30, или 60, или 90 мин.
9. Способ по любому из пп.1-8, в котором на стадии (с) матричную форму препарата с пролонгированным высвобождением нагревают до температуры по меньшей мере 60 или 62°С, но ниже 90 или ниже 80°С.
10. Способ по любому из пп.1-9, в котором стадию отверждения (c) проводят в сушильном шкафу с заданной температурой внутри шкафа.
11. Способ по любому из пп.1-9, в котором стадию отверждения (c) проводят в конвекционном аппарате для отверждения с установлением температуры воздуха на входе, на выходе и/или температуры температурного датчика.
12. Способ по любому из пп.1-9 и 11, в котором стадию отверждения (c) проводят в движущемся слое матричных форм лекарственного препарата с пролонгированным высвобождением.
13. Способ по п.12, в котором отверждение проводят в дражировочном котле.
14. Способ по любому из пп.1-13, дополнительно включающий стадию нанесения покрытия на отвержденную матричную форму препарата с пролонгированным высвобождением.
15. Способ по п.14, включающий следующие стадии:
(a) объединение (1) по меньшей мере одного полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000 и (2) по меньшей мере одного активного ингредиента с образованием композиции;
(b) формование указанной композиции в матричную форму препарата с пролонгированным высвобождением в виде таблетки прямым прессованием;
(c) отверждение указанной таблетки путем нагревания движущегося слоя таблеток до температуры от 62 до 90°С в течение по меньшей мере 1 мин в дражировочном котле и последующим охлаждением движущегося слоя таблеток до температуры ниже 50°С; затем
(d) нанесение покрытия на лекарственную форму в указанном дражировочном котле.
16. Способ изготовления твердой лекарственной формы для перорального применения с пролонгированным высвобождением, включающий, по меньшей мере, следующие стадии:
(a) объединение (1) по меньшей мере одного полиэтиленоксида, имеющего, по данным реологических измерений, молекулярную массу по меньшей мере 1000000, и (2) по меньшей мере одного активного ингредиента с образованием композиции;
(b) формование композиции с образованием матричной формы препарата с пролонгированным высвобождением;
(c) отверждение указанной матричной формы препарата с пролонгированным высвобождением, включающее, по меньшей мере, стадию отверждения, на которой указанный полиэтиленоксид, по меньшей мере, частично плавится.
17. Способ по п.16, в котором на стадии (с) по меньшей мере 20%, по меньшей мере 40% или по меньшей мере 75% высокомолекулярного полиэтиленоксида плавится.
18. Способ по п.17, в котором 100% высокомолекулярного полиэтиленоксида плавится.
19. Способ по любому из пп.1-18, в котором активным ингредиентом является опиоидный анальгетик.
20. Способ по п.19, в котором опиоидный анальгетик выбирают из группы, включающей альфентанил, аллилпродин, альфа-продин, анилеридин, бензилморфин, безитрамид, бупренорфин, буторфанол, клонитазен, кодеин, дезоморфин, декстроморамид, дезоцин, диампромид, диаморфон, дигидрокодеин, дигидроморфин, дименоксадол, димефептанол, диметилтиамбутен, диоксафетил бутират, дипипанон, эптазоцин, этогептазин, этилметилтиамбутен, этилморфин, этонитазен, эторфин, дигидроэторфин, фентанил и производные, гидрокодон, гидроморфон, гидроксипетидин, изометадон, кетобемидон, леворфанол, левофенацилморфан, лофентанил, меперидин, мептазинол, метазоцин, метадон, метопон, морфин, мирофин, нарцеин, никоморфин, норлеворфанол, норметадон, налорфин, налбуфен, норморфин, норпипанон, опиум, оксикодон, оксиморфон, папаверетум, пентазоцин, фенадоксон, феноморфан, феназоцин, феноперидин, пиминодин, пиритрамид, профептазин, промедол, проперидин, пропоксифен, суфентанил, тилидин, трамадол, их фармацевтически приемлемые соли, гидраты и сольваты, смеси любых указанных препаратов.
21. Способ по п.19, в котором опиоидный анальгетик выбирают из группы кодеина, морфина, оксикодона, гидрокодона, гидроморфона или оксиморфона или их фармацевтически приемлемых солей, гидратов и сольватов, а также смесей любых указанных препаратов.
22. Способ по п.21, в котором опиоидный анальгетик представляет собой оксикодон гидрохлорид и лекарственная форма содержит 5-500 мг оксикодон гидрохлорида.
23. Способ по п.22, в котором лекарственная форма содержит 5, 7,5, 10, 15, 20, 30, 40, 45, 60, 80, 90, 120 или 160 мг оксикодон гидрохлорида.
24. Способ по любому из пп.21-23, в котором активный ингредиент представляет собой оксикодон гидрохлорид, причем оксикодон гидрохлорид содержит 14-гидроксикодеинон в концентрации менее 25 м.д.
25. Способ по п.19, в котором опиоидный анальгетик представляет собой гидроморфон гидрохлорид и лекарственная форма содержит 1-100 мг гидроморфона гидрохлорида.
26. Способ по любому из пп.1-25, в котором по меньшей мере один полиэтиленоксид имеет молекулярную массу 2000000-8000000, по данным реологических измерений.
27. Способ по любому из пп.1-26, в котором композиция также содержит по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, менее 1000000.
28. Способ по п.27, в котором композиция также содержит по меньшей мере один полиэтиленоксид с примерной молекулярной массой, по данным реологических измерений, от 100000 до 900000.
29. Способ по п.28, в котором композиция также содержит по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, 100000.
30. Способ по любому из пп.1-29, в котором общее содержание полиэтиленоксида в композиции составляет по меньшей мере 80 мас.%.
31. Способ по любому из пп.1-30, в котором активный ингредиент представляет собой оксикодон гидрохлорид и его общее содержание в композиции составляет более 5 мас.%.
32. Способ по любому из пп.1-31, в котором содержание в композиции по меньшей мере одного полиэтиленоксида с молекулярной массой, по реологическим данным, по меньшей мере 1000000 составляет по меньшей мере 80 мас.%.
33. Способ по любому из пп.1-32, в котором композиция содержит по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000 и по меньшей мере один полиэтиленоксид с молекулярной массой, по реологическим данным, менее 1000000, причем композиция включает по меньшей мере 10 мас.% или по меньшей мере 20 мас.% полиэтиленоксида с молекулярной массой, по реологическим данным, менее 1000000.
34. Способ по любому из пп.1-33, в котором стадия отверждения (c) приводит к уменьшению плотности матричной формы лекарственного препарата с пролонгированным высвобождением.
35. Способ по п.34, в котором плотность отвержденного матричного препарата с пролонгированным высвобождением меньше плотности неотвержденной матричной формы препарата с пролонгированным высвобождением по меньшей мере на 0,5%.
36. Способ по любому из пп.1-35, в котором отверждение проводят при атмосферном давлении.
37. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением, полученная способом по любому из пп.1-36.
38. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением, включающая матричную форму препарата с пролонгированным высвобождением, включающую композицию, содержащую по меньшей мере один активный ингредиент и по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000, причем матричная форма лекарственного препарата с пролонгированным высвобождением получена отверждением при температуре по меньшей мере 60°С в течение по меньшей мере 1 мин, в форме таблетки или множества частиц, причем таблетку или множество отдельных частиц можно, по меньшей мере, сплющить без разрушения, отличающаяся толщиной таблетки или множества отдельных частиц после сплющивания, которая соответствует не более чем 60% от толщины таблетки или множества отдельных частиц до сплющивания и в которой указанная сплющенная или несплющенная таблетка либо множество сплющенных или несплющенных частиц обеспечивают такую скорость растворения in vitro при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), содержащего 40% этанол, при 37°С, которая отличается тем, что количество активного ингредиента в процентах, высвободившегося за 0,5 ч растворения, отличается не более чем на 20% пунктов от соответствующей скорости растворения in vitro, определенной на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF) и этанола, при 37°С, при использовании сплющенной или несплющенной эталонной таблетки либо множества сплющенных или несплющенных эталонных частиц соответственно.
39. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением, содержащая матричную форму препарата с пролонгированным высвобождением, которая получена отверждением при температуре по меньшей мере 60°С в течение по меньшей мере 1 мин, включающую композицию, содержащую:
(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;
(2) по меньшей мере один активный ингредиент, выбранный из опиоидных анальгетиков, и
в которой композиция содержит по меньшей мере 80 мас.% полиэтиленоксида.
40. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по п.39, в которой опиоидный анальгетик представляет собой оксикодон гидрохлорид или гидроморфон гидрохлорид и композиция содержит более 5 мас.% оксикодон гидрохлорида или гидроморфон гидрохлорида.
41. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по п.39, в которой композиция содержит по меньшей мере 80 мас.% полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000.
42. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по п.39, в которой композиция содержит по меньшей мере 10 мг оксикодон гидрохлорида и по меньшей мере 85 мас.% полиэтиленоксида.
43. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по п.39, в которой композиция содержит 15 или 20 мг оксикодон гидрохлорида.
44. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением, содержащая матричную форму препарата с пролонгированным высвобождением, которая получена отверждением при температуре по меньшей мере 60°С в течение по меньшей мере 1 мин, включающую композицию, содержащую:
(1) по меньшей мере один полиэтиленоксид с примерной молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;
(2) 40 мг оксикодон гидрохлорида, и
в которой композиция содержит по меньшей мере 65 мас.% полиэтиленоксида.
45. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением, содержащая матричную форму препарата с пролонгированным высвобождением, которая получена отверждением при температуре по меньшей мере 60°С в течение по меньшей мере 1 мин, включающую композицию, содержащую:
(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;
(2) 60 или 80 мг оксикодон гидрохлорида, и
в которой композиция содержит по меньшей мере 60 мас.% полиэтиленоксида.
46. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по п.39, в которой композиция содержит 8 мг гидроморфон гидрохлорида и по меньшей мере 94 мас.% полиэтиленоксида.
47. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по п.39, в которой композиция содержит 12 мг гидроморфон гидрохлорида и по меньшей мере 92 мас.% полиэтиленоксида.
48. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по п.39, в которой композиция содержит 32 мг гидроморфон гидрохлорида и по меньшей мере 90 мас.% полиэтиленоксида.
49. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по п.39, содержащая матричную форму препарата с пролонгированным высвобождением, включающую композицию, содержащую:
(1) по меньшей мере один активный ингредиент, который выбирают из опиоидных анальгетиков;
(2) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;
(3) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, менее 1000000.
50. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по любому из пп.37-49, в которой плотность матричной формы препарата с пролонгированным высвобождением равна или меньше 1,20 г/см3.
51. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением, содержащая матричную форму препарата с пролонгированным высвобождением, включающую композицию, содержащую:
(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;
(2) по меньшей мере один активный ингредиент,
причем матричная форма препарата с пролонгированным высвобождением получена отверждением при температуре по меньшей мере 60°С в течение по меньшей мере 1 мин и в тесте на вдавливание выдерживает усилие до растрескивания по меньшей мере 110 Н.
52. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по п.51, в которой матричная форма препарата с пролонгированным высвобождением в тесте на вдавливание характеризуется глубиной проникновения до растрескивания по меньшей мере 1,0 мм.
53. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по любому из пп.51 и 52, в которой матричная форма препарата с пролонгированным высвобождением характеризуется усилием до растрескивания по меньшей мере 120 Н, по меньшей мере 130 Н или по меньшей мере 140 Н и/или глубиной проникновения до растрескивания по меньшей мере 1,2 мм.
54. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по любому из пп.51 и 52, в которой матричная форма препарата с пролонгированным высвобождением сохраняет свою форму при нагрузке примерно 0,06 Дж без растрескивания.
55. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по любому из пп.38, 39, 42-49, 51 и 52, причем форма растворяется со скоростью, при которой при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37°С, высвобождается 12,5-55 мас.% активного ингредиента через 1 ч, 25-65 мас.% активного ингредиента через 2 ч, 45-85 мас.% активного ингредиента через 4 ч и 55-95 мас.% активного ингредиента через 6 ч.
56. Лекарственная форма препарата с пролонгированным высвобождением по пп.51-54, в которой композиция содержит по меньшей мере 80 мас.% полиэтиленоксида.
57. Лекарственная форма препарата с пролонгированным высвобождением по п.56, в которой композиция содержит по меньшей мере 80 мас.% полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000.
58. Лекарственная форма препарата с пролонгированным высвобождением по любому из пп.38-57, в которой активный ингредиент представляет собой опиоидный анальгетик.
59. Лекарственная форма препарата с пролонгированным высвобождением по п.58, в которой опиоидный анальгетик выбирают из группы, включающей альфентанил, аллилпродин, альфа-продин, анилеридин, бензилморфин, безитрамид, бупренорфин, буторфанол, клонитазен, кодеин, дезоморфин, декстроморамид, дезоцин, диампромид, диаморфон, дигидрокодеин, дигидроморфин, дименоксадол, димефептанол, диметилтиамбутен, диоксафетил бутират, дипипанон, эптазоцин, этогептазин, этилметилтиамбутен, этилморфин, этонитазен, эторфин, дигидроэторфин, фентанил и его производные, гидрокодон, гидроморфон, гидроксипетидин, изометадон, кетобемидон, леворфанол, левофенацилморфан, лофентанил, меперидин, мептазинол, метазоцин, метадон, метопон, морфин, мирофин, нарцеин, никоморфин, норлеворфанол, норметадон, налорфин, налбуфен, норморфин, норпипанон, опиум, оксикодон, оксиморфон, папаверетум, пентазоцин, фенадоксон, феноморфан, феназоцин, феноперидин, пиминодин, пиритрамид, профептазин, промедол, проперидин, пропоксифен, суфентанил, тилидин, трамадол, их фармацевтически приемлемые соли, гидраты и сольваты, смеси любых препаратов.
60. Лекарственная форма препарата с пролонгированным высвобождением по п.58, для которой опиоидный анальгетик выбирают из группы, включающей кодеин, морфин, оксикодон, гидрокодон, гидроморфон или оксиморфон или их фармацевтически приемлемые соли, гидраты и сольваты, смеси любых указанных препаратов.
61. Лекарственная форма препарата с пролонгированным высвобождением по п.58, в которой опиоидный анальгетик представляет собой оксикодон гидрохлорид и лекарственная форма содержит 5-500 мг оксикодон гидрохлорида.
62. Лекарственная форма препарата с пролонгированным высвобождением по п.58, в которой опиоидный анальгетик представляет собой гидроморфон гидрохлорида и лекарственная форма содержит 1-100 мг гидроморфон гидрохлорида.
63. Твердая лекарственная форма для перорального применения с пролонгированным высвобождением по любому из пп.38, 39, 42-49, 51, 52, в которой активный ингредиент представляет собой оксикодон гидрохлорид и в которой лекарственная форма, содержащая 10 мг оксикодон гидрохлорида, при тестировании в сравнительных клинических условиях биоэквивалентна эталонной таблетке, представляющей собой коммерческий препарат OxyContin®, содержащий 10 мг оксикодон гидрохлорида в матричной форме препарата, включающей:
a) оксикодон гидрохлорид: 10,0 мг;
b) лактоза (распылительная сушка): 69,25 мг;
c) повидон: 5,0 мг;
d) Eudragit® RS 30D (тверд.): 10,0 мг;
e) Triacetin®: 2,0 мг;
f) стеариловый спирт: 25,0 мг;
g) тальк: 2,5 мг;
h) стеарат магния: 1,25 мг; и
изготовленной следующим путем:
1) Eudragit® RS 30D и Triacetin® объединяют, пропуская через сито 60 меш, и перемешивают на низкой скорости в течение примерно 5 мин или до достижения равномерного распределения;
2) оксикодон HCl, лактозу и повидон помещают в чашку гранулятора с кипящим слоем/сушителя (FBD) и суспензию распыляют на порошок в кипящем слое;
3) если нужно уменьшить размер частиц, то после распыления гранулят пропускают через сито #12;
4) сухой гранулят помещают в миксер;
5) одновременно требуемое количество стеарилового спирта расплавляют при температуре примерно 70°С;
6) расплавленный стеариловый спирт добавляют в гранулят при перемешивании;
7) воскообразный гранулят переносят в гранулятор с кипящим слоем/сушитель и дают остыть до комнатной температуры или ниже;
8) охлажденный гранулят просеивают через сито #12;
9) воскообразный гранулят помещают в смеситель/блендер и покрывают нужными количествами талька и стеарата магния в течение примерно 3 мин;
10) гранулят прессуют в 125 мг таблетки на подходящем аппарате для таблетирования.
64. Лекарственная форма препарата с пролонгированным высвобождением по любому из пп.61, 63, которая содержит 5, 7,5, 10, 15, 20, 30, 40, 45, 60, 80, 90, 120 или 160 мг оксикодон гидрохлорида.
65. Лекарственная форма препарата с пролонгированным высвобождением по любому из пп.58 или 63, 64, в которой опиоидный анальгетик является оксикодон гидрохлоридом, содержащим 14-гидроксикодеинон в концентрации менее 25 м.д.
66. Лекарственная форма препарата с пролонгированным высвобождением по любому из пп.38-65 в форме таблетки, полученной прямым прессованием композиции и отвержденной, по меньшей мере, путем нагревания указанной таблетки до температуры по меньшей мере 60 или по меньшей мере 62°С в течение по меньшей мере 1 мин.
67. Лекарственная форма препарата с пролонгированным высвобождением по любому из пп.38-66 в виде таблетки, которая сверху покрыта слоем порошка полиэтиленоксида с образованием таблетки, содержащей внутреннюю часть таблетки и слой полиэтиленоксида, окружающий внутреннюю часть таблетки.
68. Лекарственная форма препарата с пролонгированным высвобождением по любому из пп.38-67, которая имеет форму двухслойной или многослойной таблетки, причем один из слоев содержит препарат с пролонгированным высвобождением, а один из других слоев содержит препарат немедленного высвобождения.
69. Способ лечения боли, в котором лекарственную форму по любому из пп.38-68 вводят пациенту, причем лекарственная форма содержит опиоидный анальгетик.
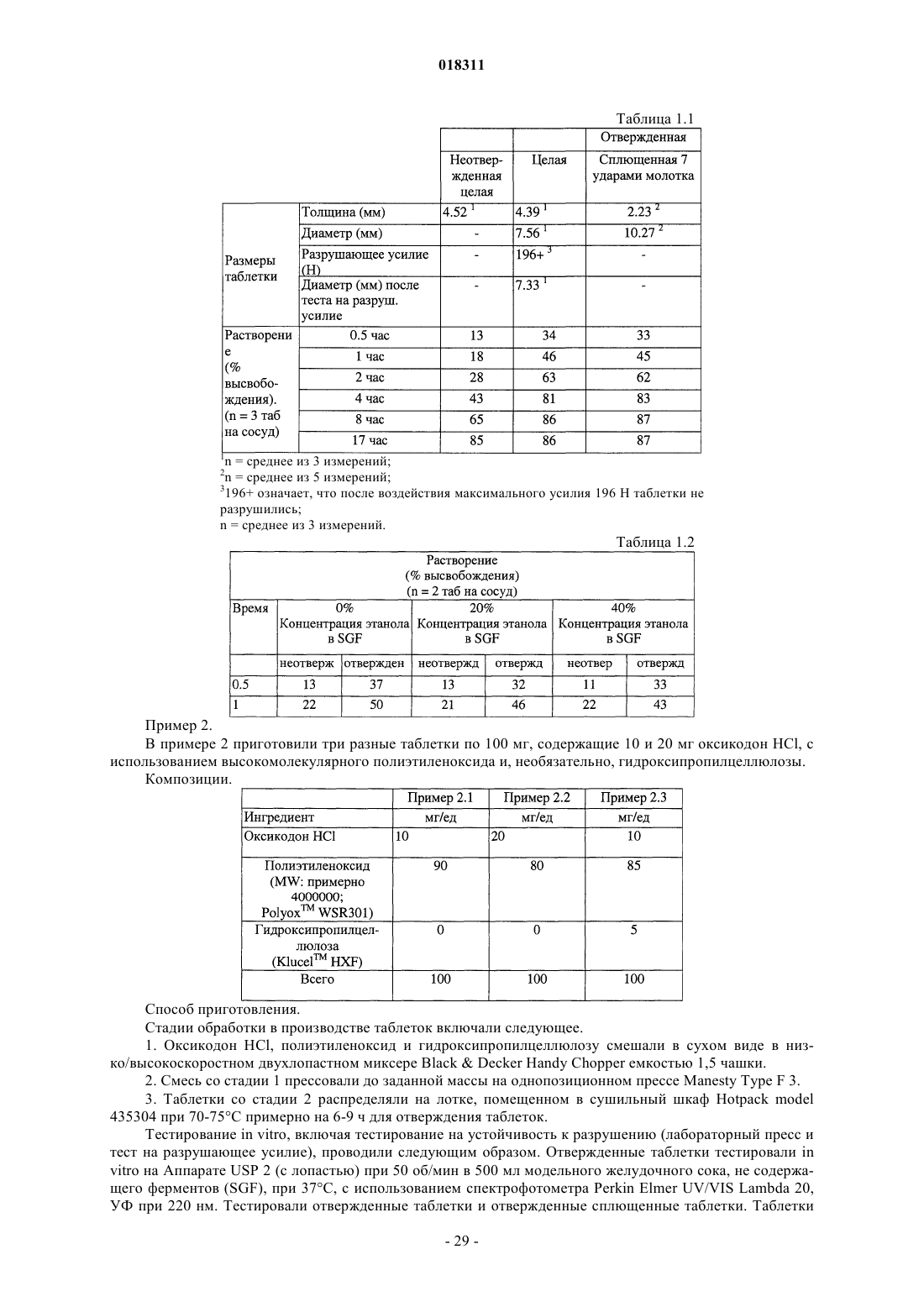
Текст
УСТОЙЧИВЫЕ ОРАЛЬНЫЕ ФАРМАЦЕВТИЧЕСКИЕ ДОЗИРОВАННЫЕ ФОРМЫ,СОДЕРЖАЩИЕ ОПИОИДНЫЙ АНАЛЬГЕТИК Изобретение относится к твердым лекарственным формам для перорального применения с пролонгированным высвобождением активного ингредиента, содержащим по меньшей мере один полиэтиленоксид, и к способам их изготовления. Раскрыт также способ лечения боли с помощью указанной лекарственной формы, содержащей опиоидный анальгетик в качестве активного ингредиента. Способ изготовления лекарственной формы включает в себя объединение полиэтиленоксида с молекулярной массой не менее 1000000 и активного ингредиента,формирование из них композиции с образованием матричной формы лекарственного препарата с пролонгированным высвобождением и отверждение указанной формы, в процессе которого форму нагревают до температуры, превышающей температуру размягчения полиэтиленоксида. Мэннион Ричард Оуэн, О'Доннэлл Эдвард Патрик, Маккенна Уильям Генри, Хуан Хайюн Хью (US) Дементьев В.Н. (RU) Область техники Настоящее изобретение относится к фармацевтическим лекарственным формам, например устойчивой к разрушению лекарственной форме, включающей опиоидный анальгетик, и способам их производства и применения, а также к методам лечения. Предпосылки создания изобретения Иногда встречаются случаи злоупотребления фармацевтическими средствами. Например, конкретная доза опиоидного агониста может действовать более эффективно при парентеральном введении, чем та же доза при пероральном употреблении. Некоторые препараты разрушают для того, чтобы использовать содержащийся в них опиоидный агонист для недозволенных целей. Например, иногда наркозависимые лица измельчают или подвергают экстракции растворителями (например, этанолом) препараты опиоидных агонистов с регулируемым высвобождением с тем, чтобы быстро выделить содержащиеся в них опиоиды для перорального или парентерального употребления. Лекарственные формы опиоидных агонистов с пролонгированным высвобождением, которые могут высвободить часть опиоида при контакте с этанолом, могут также привести к тому, что пациент получит дозу быстрее, чем это планировалось, если пациент не ознакомится с инструкцией по применению и примет алкоголь вместе с лекарственной формой. Остается потребность в разработке лекарственных форм для перорального введения, содержащих опиоидный агонист, в которых не наблюдалось бы существенного изменения высвобождения опиоида при контакте с алкоголем и/или которые обладали бы достаточной устойчивостью к разрушению. Сущность изобретения Задачей настоящего изобретения является создание устойчивой к разрушению лекарственной формы для перорального введения с пролонгированным высвобождением, содержащей активный ингредиент, такой как опиоидный анальгетик. Другой задачей настоящего изобретения является создание лекарственной формы для перорального введения с пролонгированным высвобождением, содержащей активный ингредиент типа опиоидного анальгетика, устойчивой к измельчению. Еще одной задачей настоящего изобретения является предложение лекарственной формы для перорального введения с пролонгированным высвобождением, содержащей активный ингредиент, такой как опиоидный анальгетик, устойчивой к экстракции алкоголем и потере дозы при применении его вместе со спиртом или при контакте со спиртом. В первом аспекте настоящее изобретение относится к способу изготовления твердой лекарственной формы для перорального применения с пролонгированным высвобождением, включающему, по меньшей мере, следующие стадии:(а) объединение (1) по меньшей мере одного полиэтиленоксида, имеющего, по данным реологических измерений, молекулярную массу по меньшей мере 1000000, и (2) по меньшей мере одного активного ингредиента с образованием композиции;(b) формование композиции с образованием матричной формы лекарственного препарата с пролонгированным высвобождением;(с) отверждение указанной матричной формы лекарственного препарата с пролонгированным высвобождением, включающее, по меньшей мере, стадию отверждения, на которой матричную форму препарата с пролонгированным высвобождением нагревают до температуры, которая является, по меньшей мере, температурой размягчения указанного полиэтиленоксида, в течение по меньшей мере 1 мин. В некоторых вариантах настоящее изобретение относится к способу изготовления твердой лекарственной формы для перорального применения с пролонгированным высвобождением, включающему, по меньшей мере, следующие стадии:(а) объединение (1) по меньшей мере одного полиэтиленоксида, имеющего, по данным реологических измерений, молекулярную массу по меньшей мере 1000000 и (2) по меньшей мере одного активного ингредиента с образованием композиции;(b) формование композиции с образованием матричной формы препарата с пролонгированным высвобождением;(с) отверждение указанной матричной формы препарата с пролонгированным высвобождением,включающее, по меньшей мере, стадию отверждения, на которой указанный полиэтиленоксид, по меньшей мере, частично плавится. Настоящее изобретение относится также к твердой лекарственной форме для перорального введения с пролонгированным высвобождением, полученной способами, представленными выше. Твердая лекарственная форму согласно изобретению включает матричную форму препарата с пролонгированным высвобождением, включающую композицию, содержащую по меньшей мере один активный ингредиент и по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000, причем матричная форма лекарственного препарата с пролонгированным высвобождением получена отверждением при температуре по меньшей мере 60 С в течение по меньшей мере 1 мин, в форме таблетки или множества частиц, причем таблетку или множество отдельных частиц можно, по меньшей мере, сплющить без разрушения, отличающаяся толщиной таблетки или множества отдельных частиц после сплющивания, которая соответствует не более чем 60% от толщины таблетки или множества отдельных частиц до сплющивания, и в которой указанная сплющенная таблетка или множество сплющенных частиц обеспечивают такую скорость растворения in vitro при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), содержащего 40% этанол, при 37 С, которая отличается тем, что количество активного ингредиента в процентах, высвободившегося за 0,5 ч растворения, отличается не более чем на 20% пунктов от соответствующей скорости растворения in vitro, определенной на АппаратеUSP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов(SGF) и этанола, при 37 С, при использовании сплющенной или несплющенной эталонной таблетки или множества сплющенных или несплющенных эталонных частиц соответственно. В другом варианте твердая лекарственная форма для перорального введения с пролонгированным высвобождением содержит матричную форму препарата с пролонгированным высвобождением, которая получена отверждением при температуре по меньшей мере 60 С в течение по меньшей мере 1 мин,включающую композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;(2) по меньшей мере один активный ингредиент, выбранный из опиоидных анальгетиков, и в которой композиция содержит по меньшей мере 80 мас.% полиэтиленоксида. В указанной форме в одном из вариантов опиоидный анальгетик представляет собой оксикодон гидрохлорид или гидроморфон гидрохлорид, и композиция содержит более 5 мас.% оксикодон гидрохлорида или гидроморфон гидрохлорида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального введения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, которая отверждается при температуре по меньшей мере 60 С в течение по меньшей мере 1 мин, включающую композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;(2) по меньшей мере 10 мг оксикодон гидрохлорида, и в которой композиция содержит по меньшей мере 85 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального введения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, которая отверждается при температуре по меньшей мере 60 С в течение по меньшей мере 1 мин, включающую композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;(2) по меньшей мере 15 или 20 мг оксикодон гидрохлорида; и в которой композиция содержит по меньшей мере 80 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального введения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, которая отверждается при температуре по меньшей мере 60 С в течение по меньшей мере 1 мин, включающую композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;(2) по меньшей мере 40 мг оксикодон гидрохлорида, и в которой композиция содержит по меньшей мере 65 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального введения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, которая получена отверждением при температуре по меньшей мере 60 С в течение по меньшей мере 1 мин, включающую композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;(2) по меньшей мере 60 или 80 мг оксикодон гидрохлорида, и в которой композиция содержит по меньшей мере 60 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального введения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, которая получена отверждением при температуре по меньшей мере 60 С в течение по меньшей мере 1 мин, включающую композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;(2) по меньшей мере 8 мг гидроморфон гидрохлорида, и в которой композиция содержит по меньшей мере 94 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального введения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, которая получена отверждением при температуре по меньшей мере 60 С в течение по меньшей мере 1 мин, включающую композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;(2) по меньшей мере 12 мг гидроморфон гидрохлорида, и в которой композиция содержит по меньшей мере 92 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального введения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, которая получена отверждением при температуре по меньшей мере 60 С в течение по меньшей мере 1 мин, включющую композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;(2) по меньшей мере 32 мг гидроморфон гидрохлорида, и в которой композиция содержит по меньшей мере 90 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального введения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, которая отверждается при температуре по меньшей мере 60 С в течение по меньшей мере 1 мин, включающую композицию, содержащую:(1) по меньшей мере один активный ингредиент, который выбирают из опиоидных анальгетиков;(2) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;(3) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, менее 1000000, и в которой композиция содержит по меньшей мере 80 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального введения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, включающую композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000;(2) по меньшей мере один активный ингредиент, и в которой матричная форма препарата с пролонгированным высвобождением получена отверждением при температуре по меньшей мере 60 С в течение по меньшей мере 1 мин и характеризуется в тесте на вдавливание разрушающим усилием по меньшей мере 110 Н. В некоторых вариантах матричная форма препарата с пролонгированным высвобождением в тесте на вдавливание характеризуется "глубиной проникновения до разрушения" по меньшей мере 1 мм. В некоторых вариантах настоящее изобретение относится к методу лечения, при котором лекарственную форму согласно данному изобретению, содержащую опиоидный анальгетик, назначают пациенту для болеутоления. В некоторых вариантах настоящее изобретение относится к способу получения твердой лекарственной формы для перорального введения с пролонгированным высвобождением, включающему, по меньшей мере, следующие стадии:(a) объединение (1) по меньшей мере одного полиэтиленоксида с молекулярной массой, по данным реологических измерений,по меньшей мере 1000000 и (2) по меньшей мере одного активного ингредиента с образованием композиции;(b) формование композиции с образованием матричной формы препарата с пролонгированным высвобождением;(c) отверждение указанной матричной формы препарата с пролонгированным высвобождением,включающее, по меньшей мере, стадию отверждения путем нагревания матричного препарата с пролонгированным высвобождением до температуры, которая является, по меньшей мере, температурой размягчения указанного полиэтиленоксида, в течение по меньшей мере 5 мин. Согласно некоторым вариантам изобретения твердую лекарственную форму с пролонгированным высвобождением используют в виде суппозитория. Термин "пролонгированное высвобождение" используют в целях данного изобретения для продуктов, которые служат для изготовления лекарства, которое действует в течение длительного времени после введения путем глотания, что позволяет уменьшить частоту приема формы по сравнению с традиционной лекарственной формой (например, раствором или лекарственной формой немедленного высвобождения). Термин "немедленное высвобождение" используют в данном изобретении для продуктов, которые служат для изготовления лекарства, которое растворяется в содержимом желудка без промедления или без продолжительного растворения или всасывания. Термин "твердая лекарственная форма для перорального введения с пролонгированным высвобождением" относится к вводимой форме, содержащей единичную дозу активного ингредиента в форме с пролонгированным высвобождением, такой как "матричная форма препарата с пролонгированным высвобождением", и необязательно другие адъюванты и добавки, традиционные в данной области, такие как защитное покрытие или капсулы и т.п., и необязательно любые другие добавки или компоненты, используемые в лекарственных формах. Если не оговорено особо, термин "твердая лекарственная форма для перорального введения с пролонгированным высвобождением" относится к указанной лекарственной форме в неповрежденном виде, т.е. до любого разрушения. Лекарственная форма с пролонгированным высвобождением может быть, например, в виде таблетки, содержащей лекарственную форму с пролонгированным высвобождением, или капсулы, содержащей матричную форму препарата с пролонгированным высвобождением в виде множества частиц. "Лекарственная форма с пролонгированным высвобождением" может содержать часть активного ингредиента в форме с пролонгированным высвобождением и другую часть активного ингредиента в форме с немедленным высвобождением, например в виде слоя активного ингредиента с немедленным высвобождением, окружающего лекарственную форму или компонент с немедленным высвобождением в составе лекарственной формы. Термин "матричная форма препарата с пролонгированным высвобождением" используют в целях данного изобретения для сформованной твердой формы композиции, содержащей по меньшей мере один активный ингредиент и имеющей по меньшей мере одну особенность - пролонгированное высвобождение, например, благодаря веществу матрицы для пролонгированного высвобождения, такому как высокомолекулярный полиэтиленоксид. Необязательно, композиция может содержать более двух таких соединений, а именнно другие активные ингредиенты и добавленные замедлители и/или другие вещества,включая, но не ограничиваясь ими, низкомолекулярные полиэтиленоксиды и другие адъюванты и добавки, традиционные в данной области. Термин "биоэквивалентный/биоэквивалентность" используют в целях настоящего изобретения для обозначения лекарственной формы, которая характеризуется среднегеометрическими значениями Cmax,AUCt и AUCinf для активного ингредиента, причем 90% доверительные интервалы, установленные для соотношения (тест/эталон), попадают в интервал 80-125%. Предпочтительно, чтобы средние значенияCmax, AUCt и AUCinf, определенные как для состояния после еды, так и для состояния натощак, попадали в интервал 80-125%. Термин "полиэтиленоксид" используют в целях данного изобретения для молекулярной массы по меньшей мере 25000, определенной, как принято в данной области, и предпочтительно для молекулярной массы по меньшей мере 100000. Композиции с более низкими молекулярными массами обычно определяют как полиэтиленгликоли. Термин "высокомолекулярный полиэтиленоксид" используют в целях настоящего изобретения для молекулярной массы по меньшей мере примерно 1000000. Для целей этого изобретения примерную молекулярную массу определяют реологическим методом. Считают, что полиэтиленоксид имеет молекулярную массу примерно 1000000, если 2 мас.% водный раствор указанного полиэтиленоксида имеет вязкость в интервале 400-800 мПас (сП), определенную на вискозиметре Brookfield Model RVF со шпинделем 1 при 10 об/мин и 25 С. Полиэтиленоксид имеет молекулярную массу примерно 2000000, если 2 мас.% водный раствор указанного полиэтиленоксида имеет вязкость в интервале 2000-4000 мПас (сП),определенную на вискозиметре Brookfield Model RVF со шпинделем 3 при 10 об/мин и 25 С. Полиэтиленоксид имеет молекулярную массу примерно 4000000, если 1 мас.% водный раствор указанного полиэтиленоксида имеет вязкость в интервале 1650-5500 мПас (сП), определенную на вискозиметреBrookfield Model RVF со шпинделем 2 при 2 об/мин и 25 С. Полиэтиленоксид имеет молекулярную массу примерно 5000000, если 1 мас.% водный раствор указанного полиэтиленоксида имеет вязкость в интервале 5500-7500 мПас (сП), определенную на вискозиметре Brookfield Model RVF со шпинделем 2 при 2 об/мин и 25 С. Полиэтиленоксид имеет молекулярную массу примерно 7000000, если 1 мас.% водный раствор указанного полиэтиленоксида имеет вязкость в интервале 7500-10000 мПас (сП), определенную на вискозиметре Brookfield Model RVF со шпинделем 2 при 2 об/мин и 25 С. Полиэтиленоксид имеет молекулярную массу примерно 8000000, если 1 мас.% водный раствор указанного полиэтиленоксида имеет вязкость в интервале 10000-15000 мПас (сП), определенную на вискозиметре Brookfield Model RVF со шпинделем 2 при 2 об/мин и 25 С. Для низкомолекулярных полиэтиленоксидов принято считать, что полиэтиленоксид имеет молекулярную массу примерно 100000, если 5 мас.% водный раствор указанного полиэтиленоксида имеет вязкость в интервале 30-50 мПас (сП), определенную на вискозиметре Brookfield Model RVF со шпинделем 1 при 50 об/мин и 25 С и полиэтиленоксид имеет молекулярную массу примерно 900000, если 5 мас.% водный раствор указанного полиэтиленоксида имеет вязкость в интервале 8800-17600 мПас (сП), определенную на вискозиметре Brookfield Model RVF со шпинделем 2 при 2 об/мин и 25 С. Термин "низкомолекулярный полиэтиленоксид" используют в целях данного изобретения для молекулярной массы менее примерно 1000000, определенной описанным выше реологическим методом. Термин "прямое прессование" в целях настоящего изобретения относят к способу таблетирования, в котором таблетку или любую другую спрессованную лекарственную форму получают способом, включающим стадии сухого смешения соединений и прессования сухой смеси с образованием лекарственной формы, например с использованием диффузной смеси и/или конвекционного способа смешения (см. например, Guidance for Industry, SUPAC-IR/MR: Immediate Release and Modified Release Solid Oral DosageForms (Твердые лекарственные формы для перорального введения с немедленным и модифицированным высвобождением), Manufacturing Equipment Addendum). Термин "слой движущихся частиц" относят в целях настоящего изобретения к порции таблеток, которые движутся относительно друг друга, как, например, в дражировочных котлах, при соответствующей скорости вращения или в кипящем слое таблеток. Предпочтительно, чтобы слой свободно движущихся таблеток уменьшал слипание таблеток друг с другом или препятствовал ему. Термин "сплющивание" и родственные термины, использованные в контексте сплющивания таблеток или других лекарственных форм согласно настоящему изобретению, означают, что к таблетке прилагают усилие в направлении, практически перпендикулярном к ее диаметру и практически вдоль оси, например, таблетки. Усилие можно приложить с помощью настольного пресса (если точно не назван другой) в такой степени, которая необходима для достижения заданного соотношения сплющенности/уменьшенной толщины. В некоторых вариантах изобретения сплющивание не приводит к разламыванию таблетки на куски, однако ребра растрескиваются с образованием отдельных кусков. Сплющенность описывают в терминах толщины сплющенной таблетки по сравнению с толщиной несплющенной таблетки в % толщины по отношению к толщине несплющенной таблетки. Помимо таблеток сплющивание можно применять к любой конфигурации лекарственной формы, причем усилие прилагают в одном направлении практически вдоль линии наименьшего диаметра (т.е. вдоль оси) частицы, если частица имеет несферическую форму, и в любом направлении, если частица является сферической. Затем сплющенность описывают в терминах соотношения толщина/наименьший диаметр сплющенной конфигурации по сравнению с соотношением толщина/наименьший диаметр несплющенной конфигурации, выраженной в % толщины относительно соотношения толщина/наименьший диаметр несплющенной конфигурации, если начальная форма является несферической, или в % толщины по отношению к диаметру несплющенной частицы, если начальная форма является сферической. Толщину измеряют с помощью прибора для измерения толщины (например, цифрового толщиномера или цифрового кронциркуля). На фиг. 4-6 показаны таблетки, которые были сплющены с помощью настольного пресса. Начальная конфигурация таблеток показана на фиг. 1-3 на левой фотографии. В некоторых вариантах изобретения помимо использования настольного пресса для сплющивания таблеток/лекарственных форм можно применить молоток. При таком способе сплющивания ударяют молотком в направлении практически по линии, направленной вдоль оси, например, таблетки. Сплющенность также описывают в терминах соотношения толщина/наименьший диаметр сплющенной конфигурации по сравнению с несплющенной конфигурацией, выраженного в % толщины по отношению к соотношению толщина/наименьший диаметр для несплющенной конфигурации, если начальная форма является несферической, или в % толщины по отношению к диаметру несплющенной конфигурации,если начальная форма является сферической. Толщину измеряют с помощью прибора для измерения толщины (например, цифрового толщиномера или цифрового кронциркуля). Альтернативно тесты на разрушающее усилие или твердость таблетки можно проводить так, как это описано в Remington's Pharmaceutical Sciences, 18th edition, 1990, Chapter 89 "Oral Solid Dosage Forms",pages 1633-1665, который включен здесь ссылкой. При этом используют аппарат Schleuniger, в котором таблетку/лекарственную форму помещают между парой плоскопараллельных пластин и прессуют с помощью плоских пластин таким образом, что усилие прилагается практически перпендикулярно толщине и практически вдоль оси таблетки, что приводит к уменьшению диаметра в этом направлении. Этот уменьшенный диаметр описывают в терминах % диаметра в расчете на диаметр таблетки до проведения теста на разрушающее усилие. Разрушающее усилие или твердость таблетки определяют как усилие, при котором тестированная таблетка/лекарственная форма ломается. Таблетки/лекарственные формы, которые не ломаются, а только деформируются под действием приложенной силы, считают устойчивыми к разрушению под действием этой конкретной силы. Следующий тест для количественной оценки прочности таблеток/лекарственных форм представляет собой тест на вдавливание с использованием текстурного анализатора, такого как TA-ХТ 2 TextureAnalyzer (Texture Technologies Corp., 18 Fairview Road, Scarsdale, NY 10583). В этом способе таблетки/лекарственные формы кладут на наковальню из нержавеющей стали со слегка вогнутой поверхностью и последовательно подвергают действию падающего зонда от текстурного анализатора TA-8 А, например зонда в виде стальных шариков диаметром 1/8 дюйма. Перед началом эксперимента таблетку располагают прямо под зондом так, чтобы падающий зонд проникал в таблетку вдоль оси, т.е. в центре таблетки, а усилие от падающего зонда было приложено практически перпендикулярно диаметру и практически вдоль оси таблетки. Вначале зонд текстурного анализатора перемещается по направлению к испытуемой таблетке с предтестовой скоростью. При контакте зонда с поверхностью таблетки и при достижении усилия срабатывания зонд начинает двигаться с тестовой скоростью и проникает внутрь таблетки. Для каждой глубины проникновения пробы, которую далее будут называть "расстоянием", измеряют соответствующее усилие и суммируют данные. Когда зонд достигает нужной максимальной глубины проникновения, он изменяет направление движения и поднимается обратно с посттестовой скоростью, причем все последующие данные можно суммировать. Разрушающее усилие определяют как усилие в первом локальном максимуме, которое достигается на соответствующей диаграмме усилие/расстояние, и рассчитывают с помощью программы анализатора текстуры "Texture Expert Exceed, Version 2.64 English".He связывая себя какой-либо теорией, можно считать, что в этой точке происходит структурное повреждение таблетки/лекарственной формы в виде разрушения. Однако разрушенные таблетки/лекарственные формы, согласно некоторым вариантам настоящего изобретения, остаются целыми, о чем свидетельствует наличие устойчивости к падающему зонду. Соответствующее расстояние в первом локальном максимуме далее называют расстоянием "глубины проникновения до разрушения". Для целей некоторых вариантов настоящего изобретения термин "прочность при разрушении" относится к твердости таблеток/лекарственных форм, которую предпочтительно определять с помощью аппарата Schleuniger, а термин "разрушающее усилие" отражает прочность таблеток/лекарственных форм, которую предпочтительно определять в тесте на вдавливание с помощью текстурного анализатора. Следующим параметром матричной формы препарата с пролонгированным высвобождением, который можно оценить по результатам описанного выше теста на вдавливание, является нагрузка на матричную форму препарата с пролонгированным высвобождением в тесте на вдавливание, как описано выше. Величина нагрузки соответствует интегральному усилию на все расстояние. Термин "устойчивый к разрушению" для целей некоторых вариантов настоящего изобретения относят к лекарственным формам, которые можно, по меньшей мере, сплющить с помощью настольного пресса, как описано выше, без разрушения не более чем примерно на 60% толщины, более предпочтительно не более чем примерно на 50% толщины, более предпочтительно не более чем примерно на 40% толщины, даже более предпочтительно не более чем примерно на 30% толщины и наиболее предпочтительно не более чем примерно на 20% толщины, 10% толщины или 5% толщины. Для целей некоторых вариантов настоящего изобретения лекарственные формы считают "устойчивыми к экстракции спиртом", если соответствующие лекарственные формы обеспечивают такую скорость растворения in vitro, определенную на USP Аппарате 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока без ферментов (SGF) с 40% этанолом при 37 С, что процент активного вещества, выделившегося за 0,5 ч, предпочтительно за 0,5 и 0,75 ч, более предпочтительно за 0,5, 0,75 и 1 ч,даже более предпочтительно за 0,5, 0,75, 1 и 1,5 ч и наиболее предпочтительно за 0,75, 1, 1,5 и 2 ч растворения, отличается не более чем на 20% пунктов или предпочтительно не более чем примерно на 15% пунктов в каждый из указанных отрезков времени от соответствующей скорости растворения in vitro,определенной на USP Аппарате 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока без ферментов (SGF) при 37 С, без этанола. Термин "устойчивость к разрушению" для целей настоящего изобретения относится к лекарственным формам, которые, по меньшей мере, устойчивы к разрушению или устойчивы к экстракции спиртом, предпочтительно к тому и другому, как описано выше, и могут иметь другие параметры устойчивости. Для целей настоящего изобретения термин "активный ингредиент" относят к фармацевтически активному веществу, которое включает все без ограничения опиоидные анальгетики. Для целей настоящего изобретения термин "опиоидный анальгетик" включает отдельные соединения или композиции соединений, которые выбирают из группы опиоидов и которые оказывают анальгезирующий эффект. К их числу относятся один опиоидный агонист, или комбинация опиоидных агонистов, один смешанный опиоидный агонист-антагонист, или комбинация смешанных опиоидных агонистов-антагонистов, или часть опиоидных агонистов и одного или нескольких опиоидных антагонистов, а также стереоизомеры, простые или сложные эфиры, соли, их гидраты и сольваты, композиции из любых указанных выше соединений и т.п. Описанное здесь изобретение конкретно относится к использованию опиоидных анальгетиков в форме любых фармацевтически приемлемых солей. Фармацевтически приемлемые соли включают, но не ограничиваются ими, соли неорганических кислот, такие как гидрохлорид, гидробромид, сульфат, фосфат и т.п.; соли органических кислот, такие как формиат, ацетат, трифторацетат, малеат, тартрат и т.п.; сульфонаты, такие как метансульфонат, бензолсульфонат, п-толуолсульфонат и т.п.; соли аминокислот, такие как аргинат, аспарагинат, глютамат и т.п., и соли металлов, такие как натриевая соль, калиевая соль, цезиевая соль и т.п.; щелочно-земельных металлов, такие как кальциевая соль, магниевая соль и т.п.; соли органических аминов, такие как соль триэтиламина, соль пиридина, соль пиколина, соль этаноламина, соль триэтаноламина, соль дициклогексиламина, соль N,N'-дибензилэтилендиамина и т.п. Опиоиды, используемые согласно настоящему изобретению, могут содержать один или несколько асимметрических центров и могут включать энантиомеры, диастереомеры или другие стереоизомерные формы. Настоящее изобретение также предполагает включить использование всех возможных форм, а также их рацемических и расщепленных форм и их смесей. В случае, если описанные здесь соединения содержат олефиновые двойные связи или другие центры геометрической асимметрии, то будут включены как Е, так и Z геометрические изомеры. Все таутомеры будут также включены в настоящее изобретение. Использованный здесь термин "стереомеры" является общим термином для всех изомеров индивидуальных молекул, которые отличаются только пространственной ориентацией их атомов. Он включает энантиомеры и изомеры соединений, содержащих более одного хирального центра, которые не являются зеркальным отображением друг друга (диастереоизомеры). Термин "хиральный центр" относится к атому углерода, с которым соединены четыре разные группы. Термин "энантиомер" или "энантиомерный" относится к молекуле, которая не совпадает с ее зеркальным изображением и, следовательно, оптически активна, причем энантиомер вращает плоскость поляризованного света в одном направлении, а его зеркальное отображение вращает плоскость поляризованного света в противоположном направлении. Термин "рацемический" относится к оптически не активной смеси равных частей энантиомеров. Термин "расщепление" относится к разделению, или концентрированию, или удалению одной из двух энантиомерных форм молекулы. Опиоидные агонисты, используемые в настоящем изобретении, включают, но не ограничиваются ими, альфентанил, аллилпродин, альфа-продин, анилеридин, бензилморфин, безитрамид, бупренорфин,буторфанол, клонитазен, кодеин, дезоморфин, декстроморамид, дезоцин, диампромид, диаморфон, дигидрокодеин, дигидроморфин, дименоксадол, димефептанол, диметилтиамбутен, диоксафетил бутират,дипипанон, эптазоцин, этогептазин, этилметилтриамбутен, этилморфин, этонитазен, эторфин, дигидроэторфин, фентанил и производные, гидрокодон, гидроморфон, гидроксипетидин, изометадон, кетобемидон, леворфанол, левофенацилморфан, лофентанил, меперидин, мептазинол, метазоцин, метадон, метопон, морфин, мирофин, нарцеин, никоморфин, норлеворфанол, норметадон, налорфин, налбуфен, норморфин, норпипанон, опиум, оксикодон, оксиморфон, папаверетум, пентазоцин, фенадоксон, феноморфан, феназоцин, феноперидин, пиминодин, пиритрамид, профептазин, промедол, проперидин, пропоксифен, суфентанил, тилидин, трамадол, их фармацевтически приемлемые соли, гидраты и сольваты, смеси любых препаратов и т.п. Опиоидные антагонисты, используемые в комбинации с опиоидными агонистами, как описано выше, включают налоксон, налтрексон и налмефен или их фрамацевтически приемлемые соли, гидраты и сольваты, смеси любых указанных соединений и т.п. В некоторых вариантах используют, например, комбинацию оксикодона HCl и налоксона HCl в соотношении 2:1. В некоторых вариантах опиоидные анальгетики выбирают из кодеина, морфина, оксикодона, гидрокодона, гидроморфона или оксиморфона или их фармацевтически приемлемых солей, гидратов и сольватов, смесей любых указанных препаратов и т.п. В некоторых вариантах опиоидный анальгетик представляет собой оксикодон, гидроморфон или оксиморфон или их соль, например гидрохлорид. Лекарственная форма содержит примерно 5-500 мг оксикодон гидрохлорида, примерно 1-100 мг гидроморфон гидрохлорида или примерно 5-500 мг оксиморфон гидрохлорида. При использовании других солей, производных или форм можно использовать эквимолярные количества любых других фармацевтически приемлемых солей или производных или форм, включая, но не ограничиваясь ими, гидраты и сольваты или свободные основания. Лекарственная форма содержит, например, 5, 7,5, 10, 15, 20, 30, 40, 45, 60, 80, 90, 120 или 160 мг оксикодон гидрохлорида или эквимолярные количества любой другой фармацевтически приемлемой соли, производного или формы, включая, но не ограничиваясь ими, гидраты и сольваты или свободные основания. Лекарственная форма содержит, например, 5, 7,5, 10, 15, 20, 30, 40, 45, 60, 80, 90, 120 или 160 мг оксиморфон гидрохлорида или эквимолярные количества любой другой фармацевтически приемлемой соли, производного или формы, включая, но не ограничиваясь ими, гидраты и сольваты или свободные основания. Лекарственная форма содержит, например, 2, 4, 8, 12, 16, 24, 32, 48 или 64 мг гидроморфон гидрохлорида или эквимолярные количества любой другой фармацевтически приемлемой соли, производного или формы,включая, но не ограничиваясь ими, гидраты и сольваты или свободные основания. В патентах WO 2005/097801 А 1, США 7129248 В 2 и США 2006/0173029 А 1, которые все включены здесь ссылками, описан способ получения оксикодон гидрохлорида с концентрацией 14-гидроксикодеинона менее примерно 25 м.д., предпочтительно менее примерно 15 м.д., менее примерно 10 м.д. или менее примерно 5 м.д., более предпочтительно менее примерно 2 м.д., менее примерно 1 м.д., менее примерно 0,5 м.д. или менее примерно 0,25 м.д. Использованный здесь термин "м.д." означает "миллионная доля". Относительно 14-гидроксикодеинона "м.д." означает миллионную долю 14-гидроксикодеинона в конкретном образце. Концентрацию 14-гидроксикодеинона можно определить любым способом, известным в данной области,предпочтительно методом ВЭЖХ с УФ-детектированием. В некоторых вариантах настоящего изобретения, в которых активный ингредиент представляет собой оксикодон гидрохлорид,используют оксикодон гидрохлорид с концентрацией 14-гидроксикодеинона менее примерно 25 м.д., предпочтительно менее примерно 15 м.д., менее примерно 10 м.д. или менее примерно 5 м.д., более предпочтительно менее примерно 2 м.д., менее примерно 1 м.д., менее примерно 0,5 м.д. или менее примерно 0,25 м.д. В некоторых других вариантах согласно данному изобретению можно использовать другие терапевтически активные ингредиенты либо в комбинации с опиоидами, либо вместо опиоидов. Примеры таких терапевтически активных ингредиентов включают антигистаминовые препараты (например, дименгидринат, дифенгидрамин, хлорфенирамин и дексхлорфенирамин малеат), нестероидные противовоспалительные ингредиенты (например, напроксин, диклофенак, индометацин, ибупрофен, сулиндак,ингибиторы Кокс-2) и ацетаминофен, противорвотные средства (например, метохлопрамид, метилналтрексон), антиэпилептики (например, фенитоин, мепробамат и нитразепам), сосудорасширяющие средства (например, нифедипин, папаверин, дилтиазем и никардипин), противокашлевые и отхаркивающие средства (например, кодеин фосфат), антиастматические средства (например, теофиллин), антациды,противоспазматические средства (например, атропин, скополамин), антидиабетические средства (например, инсулин), диуретики (например, этакриновая кислота, бендрофлутиазид), антигипотензивные средства (например, пропранолол, клонидин), антигипертензивные средства (например, клонидин, метилдопа), бронхорасширяющие средства (например, албутерол), стероиды (например, гидрокортизон, триамцинолон, преднизон), антибиотики (например, тетрациклин), противогеморроидальные средства, гипнотики, психотропные, антидиарейные, миколитики, седативные, противоотечные средства (например,псевдоэфедрин), слабительные, витамины, стимуляторы (включая подавляющие аппетит типа фенилпропаноламина) и каннабиоиды, а также их фармацевтически приемлемые соли, гидраты и сольваты. В некоторых вариантах изобретение относится к использованию в качестве активных ингредиентов ингибиторов Кокс-2 в комбинации с опиоидными анальгетиками или вместо опиоидных анальгетиков,например, ингибиторов Кокс-2, таких как мелоксикам (4-гидрокси-2-метил-N-(5-метил-2-тиазолил)-2 Н 1,2-бензотиазин-3-карбоксамид-1,1-диоксид), как описано в патенте США 10/056347 и 11/825938, который включен здесь ссылкой, набуметон (4-(6-метокси-2-нафтил)-2-бутанон), как описано в патенте США 10/056348, который включен ссылкой, целекоксиб (4-[5-(4-метилфенил)-3-(трифторметил)-1 Нпиразол-1-ил]бензолсульфонамид), как описано в патенте США 11/698394, который включен здесь ссылкой, нимесулид (N-(4-нитро-2-феноксифенил)метансульфонамид), как описано в патенте США 10/057630, который включен здесь ссылкой, и N-[3-(формиламино)-4-оксо-6-фенокси-4 Н-1 бензопиран-7-ил]метансульфонамид (Т-614), как описано в патенте США 10/057632, который включен здесь ссылкой. Настоящее изобретение также относится к лекарственным формам, в которых используют такие активные ингредиенты как, например, бензодиазепины, барбитураты или амфетамины. Их можно объединять с соответствующими антагонистами. Термин "бензодиазепины" относится к бензодиазепинам и лекарствам, которые являются производными бензодиазепина и способны подавлять центральную нервную систему. Бензодиазепины включают,но не ограничиваются ими, альпразолам, бромазепам, хлордиазепоксид, хлоразепат, диазепам, эстазолам,флуразепам, халазепам, кетазолам, лоразепам, нитразепам, оксазепам, празепам, квазепам, темазепам,триазолам, метилфенидат, а также их фармацевтически приемлемые соли, гидраты и сольваты и смеси. Антагонисты бензодиазепина, которые можно использовать в настоящем изобретении, включают, но не ограничиваются ими, флумазенил, а также его фармацевтически приемлемые соли, гидраты и сольваты. Барбитураты относятся к седативно-гипнотическим лекарствам, полученным из барбитуровой кислоты (2,4,6,-триоксогексагидропиримидин). Барбитураты включают, но не ограничиваются ими, амобарбитал, апробарботал, бутабарбитал, буталбитал, метогекситал, мефобарбитал, метарбитал, пентобарбитал, фенобарбитал, секобарбитал, а также смеси их фармацевтически приемлемых солей, гидратов и сольватов. Антагонисты барбитуратов, которые можно использовать в данном изобретении, включают,но не ограничиваются ими, амфетамины, а также их фармацевтически приемлемые соли, гидраты и сольваты. Стимуляторы относятся к лекарствам, которые стимулируют центральную нервную систему. Стимуляторы включают, но не ограничиваются ими, амфетамины, такие как амфетамин, комплекс декстроамфетаминовой смолы, декстроамфетамин, метамфетамин, метилфенидат, а также их фармацевтически приемлемые соли, гидраты и сольваты и их смеси. Антагонисты стимуляторов, которые можно использовать в данном изобретении, включают, но не ограничиваются ими, бензодиазепины, а также описанные здесь их фармацевтически приемлемые соли, гидраты и сольваты. Краткое описание чертежей Фиг. 1 представляет собой вид сверху (вдоль оси таблетки) таблеток из примера 7.1 до (слева) и после (справа) теста на прочность к разрушению на аппарате Schleuniger Model 6D. Фиг. 2 представляет собой вид сверху (вдоль оси таблетки) таблеток из примера 7.2 до (слева) и после (справа) теста на прочность к разрушению на аппарате Schleuniger Model 6D. Фиг. 3 представляет собой вид сверху (вдоль оси таблетки) таблеток из примера 7.3 перед (слева) и после (справа) после теста на прочность к разрушению на аппарате Schleuniger Model 6D. Фиг. 4 представляет собой вид сверху (вдоль оси таблетки) таблетки из примера 7.1 после сплющивания с помощью настольного лабораторного пресса Carver (модель 3912). Фиг. 5 представляет собой вид сверху (вдоль оси таблетки) таблетки из примера 7.2 после сплющивания с помощью настольного лабораторного пресса Carver (модель 3912). Фиг. 6 представляет собой вид сверху (вдоль оси таблетки) таблетки из примера 7.3 после сплющивания с помощью настольного лабораторного пресса Carver (модель 3912). Фиг. 7 представляет собой вид сверху (вдоль оси таблетки) таблетки из примера 7.1 после 10 ударов ручным молотком. Фиг. 8 представляет собой вид сверху (вдоль оси таблетки) таблетки из примера 7.2 после 10 ударов ручным молотком. Фиг. 9 представляет собой вид сверху (вдоль оси таблетки) таблетки из примера 7.3 после 10 ударов ручным молотком. Фиг. 10 представляет собой диаграмму, изображающую температурный профиль способа отверждения из примера 13.1. Фиг. 11 представляет собой диаграмму, изображающую температурный профиль способа отверждения из примера 13.2. Фиг. 12 представляет собой диаграмму, изображающую температурный профиль способа отверждения из примера 13.3. Фиг. 13 представляет собой диаграмму, изображающую температурный профиль способа отверждения из примера 13.4. Фиг. 14 представляет собой диаграмму, изображающую температурный профиль способа отверждения из примера 13.5. Фиг. 15 представляет собой диаграмму, изображающую температурный профиль способа отверждения из примера 14.1. Фиг. 16 представляет собой диаграмму, изображающую температурный профиль способа отверждения из примера 14.2. Фиг. 17 представляет собой диаграмму, изображающую температурный профиль способа отверждения из примера 14.3. Фиг. 18 представляет собой диаграмму, изображающую температурный профиль способа отверждения из примера 14.4. Фиг. 19 представляет собой диаграмму, изображающую температурный профиль способа отверждения из примера 14.5. Фиг. 20 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 13.1 (отверждали в течение 30 мин, без покрытия). Фиг. 21 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 13.2 (отверждали в течение 30 мин, без покрытия). Фиг. 22 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 13.3 (отверждали в течение 30 мин, без покрытия). Фиг. 23 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 13.4 (отверждали в течение 30 мин, без покрытия). Фиг. 24 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 13.5 (отверждена в течение 30 мин, без покрытия). Фиг. 25 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 17.1 (отверждена в течение 15 мин при 72 С, с покрытием). Фиг. 26 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 18.2 (отверждена в течение 15 мин при 72 С, с покрытием). Фиг. 27 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 14.1 (отверждена в течение 1 ч, с покрытием). Фиг. 28 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 14.2 (отверждена в течение 1 ч, с покрытием). Фиг. 29 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 14.3 (отверждена в течение 1 ч, с покрытием). Фиг. 30 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 14.4 (отверждена в течение 1 ч, с покрытием). Фиг. 31 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 14.5 (отверждена в течение 1 ч, с покрытием). Фиг. 32 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 16.1 (отверждена в течение 15 мин, с покрытием). Фиг. 33 представляет собой диаграмму теста на вдавливание из примера 20 для таблетки из примера 16.2 (отверждена в течение 15 мин, с покрытием). Фиг. 34 представляет собой диаграмму теста на вдавливание из примера 21 для таблетки из примера 16.1 (отверждена в течение 15 мин, с покрытием) и для 60 мг таблетки промышленного OxyContin. Фиг. 35 представляет собой диаграмму теста на вдавливание из примера 21 для таблетки из примера 16.2 (отверждена в течение 15 мин, с покрытием) и для 80 мг таблетки промышленного OxyContin. На фиг. 36 показана средняя концентрация оксикодона в плазме в зависимости от временного профиля в линейном масштабе [совокупность: полный анализ, статус (после еды)] согласно примеру 26. На фиг. 37 показана средняя концентрация оксикодона в плазме в зависимости от временного профиля в логарифмическом масштабе [совокупность: полный анализ, статус (после еды)] согласно примеру 26. На фиг. 38 показана средняя концентрация оксикодона в плазме в зависимости от временного профиля в линейном масштабе [совокупность: полный анализ, статус (после еды)] согласно примеру 26. На фиг. 39 показана средняя концентрация оксикодона в плазме в зависимости от временного профиля в логарифмическом масштабе [совокупность: полный анализ, статус (после еды)] согласно примеру 26. На фиг. 40 показан типичный вид разрушенных таблеток OxyContin 10 мг и таблеток из примера 7.2 согласно примеру 27. На фиг. 41 показан типичный вид размолотых 10 мг таблеток из примера 7.2 и таблетокOxyContin до и после растворения в течение 45 мин согласно примеру 27. На фиг. 42 показаны профили растворения размолотой 10 мг таблетки из примера 7.2 и таблеток разрушенного OxyContin 10 мг согласно примеру 27. На фиг. 43 показаны графики распределения частиц по размерам в размолотых таблетках (таблеткиOxyContin 10 мг из примеров 7.2 и примера 14.5) согласно примеру 27. Подробное описание В некоторых вариантах настоящее изобретение относится к способу получения твердой фармацевтической лекарственной формы для перорального введения с пролонгированным высвобождением,включающему, по меньшей мере, стадии:(a) объединение (1) по меньшей мере одного полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000 и (2) по меньшей мере одного активного ингредиента с образованием композиции;(b) формование композиции с образованием матричной формы препарата с пролонгированным высвобождением;(c) отверждение указанной матричной формы препарата с пролонгированным высвобождением,включающее, по меньшей мере, стадию отверждения путем нагревания матричной формы препарата с пролонгированным высвобождением до температуры, которая является, по меньшей мере, температурой размягчения указанного полиэтиленоксида, в течение по меньшей мере примерно 1 мин. Предпочтительно проводить отверждение при атмосферном давлении. В одном варианте настоящее изобретение относится к способу получения твердой лекарственной формы для перорального введения с пролонгированным высвобождением, включающему, по меньшей мере, стадии:(а) объединение (1) по меньшей мере одного полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000 и (2) по меньшей мере одного активного ингредиента с образованием композиции;(b) формование композиции с образованием матричной формы препарата с пролонгированным высвобождением;(с) отверждение указанной матричной формы препарата с пролонгированным высвобождением,включающее, по меньшей мере, стадию отверждения путем нагревания матричной формы препарата с пролонгированным высвобождением до температуры, которая является, по меньшей мере, температурой размягчения указанного полиэтиленоксида, в течение по меньшей мере примерно 5 мин. Предпочтительно проводить отверждение при атмосферном давлении. В некоторых вариантах настоящее изобретение относится к способу получения твердой лекарственной формы для перорального введения с пролонгированным высвобождением, включающему, по меньшей мере, стадии:(а) объединение (1) по меньшей мере одного полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000 и (2) по меньшей мере одного активного ингредиента с образованием композиции;(b) формование композиции с образованием матричной формы препарата с пролонгированным высвобождением;(с) отверждение указанной матричной формы препарата с пролонгированным высвобождением,включающее, по меньшей мере, стадию отверждения, на которой указанный полиэтиленоксид, по меньшей мере, частично плавится. Предпочтительно проводить отверждение при атмосферном давлении. В некоторых вариантах композицию формуют на стадии (b) в матричную форму препарата с пролонгированным высвобождением в виде таблеток. Для формования матричной формы препарата с пролонгированным высвобождением в виде таблетки можно использовать прямое прессование. Прямое прессование является эффективным и простым способом формования таблеток, при котором можно избежать таких стадий способа, как влажная грануляция. Однако можно использовать и другие способы получения таблеток, известные в данной области, такие как влажная грануляция и последующее прессование гранул в форме таблеток. В одном варианте отверждение матричной формы препарата с пролонгированным высвобождением на стадии (c) включает, по меньшей мере, стадию отверждения, на которой высокомолекулярный полиэтиленоксид в матричной форме препарата с пролонгированным высвобождением, по меньшей мере,частично плавится. Например, плавится по меньшей мере примерно 20% или по меньшей мере примерно 30% высокомолекулярного полиэтиленоксида в матричной форме препарата с пролонгированным высвобождением. Предпочтительно, чтобы плавилось по меньшей мере примерно 40% или по меньшей мере примерно 50%, более предпочтительно по меньшей мере примерно 60%, по меньшей мере примерно 75% или по меньшей мере примерно 90% высокомолекулярного полиэтиленоксида в матричной форме препарата с пролонгированным высвобождением. В предпочтительном варианте плавится примерно 100% высокомолекулярного полиэтиленоксида. В других вариантах отверждение матричной формы препарата с пролонгированным высвобождением на стадии (c) включает, по меньшей мере, стадию отверждения, на которой матричную форму препарата с пролонгированным высвобождением нагревают до повышенной температуры в течение определенного периода времени. В таких вариантах температуры, используемые на стадии (с), т.е. температуры отверждения, достигают, по меньшей мере, температуры размягчения высокомолекулярного полиэтиленоксида. Не связывая себя какой-либо теорией, можно полагать, что отверждение при температуре, которая близка к температуре размягчения высокомолекулярного полиэтиленоксида, вызовет, по меньшей мере, прилипание частиц полиэтиленоксида друг к другу или даже их слипание. Согласно вариантам температура отверждения составляет по меньшей мере примерно 60 С, или по меньшей мере примерно 62 С, или примерно 62-90 С, или примерно 62-85 С, или примерно 62-80 С, или примерно 6590 С, или примерно 65-85 С, или примерно 65-80 С. Предпочтительно, чтобы температура отверждения находилась в интервале примерно 68-90 С, или примерно 68-85 С, или примерно 68-80 С, более предпочтительно примерно 70-90 С, или примерно 70-85 С, или примерно 70-80 С, наиболее предпочтительно примерно 72-90 С, или примерно 72-85 С, или примерно 72-80 С. Температура отверждения может быть по меньшей мере примерно 60 С или по меньшей мере 62 С, но менее примерно 90 С или менее примерно 80 С. Предпочтительно, чтобы она находилась в интервале примерно 62-72 С, в частности примерно 68-72 С. Предпочтительно, чтобы температура отверждения была равна, по меньшей мере,нижнему пределу интервала температур размягчения высокомолекулярного полиэтиленоксида или по меньшей мере примерно 62-68 С. Более предпочтительно, чтобы температура отверждения была внутри интервала температур размягчения высокомолекулярного полиэтиленоксида или по меньшей мере примерно 70 С. Даже более предпочтительно, чтобы температура отверждения была равна, по меньшей мере, верхнему пределу интервала температур размягчения высокомолекулярного полиэтиленоксида или по меньшей мере примерно 72 С. В альтернативном варианте температура отверждения может быть выше верхнего предела интервала размягчения высокомолекулярного полиэтиленоксида, например температура отверждения составляет по меньшей мере примерно 75 С или по меньшей мере примерно 80 С. В тех вариантах, в которых отверждение матричной формы препарата с пролонгированным высвобождением на стадии (c) включает, по меньшей мере, стадию отверждения, на которой матричную форму препарата с пролонгированным высвобождением нагревают до повышенной температуры в течение определенного периода времени, этот период времени далее называют временем отверждения. Для определения времени отверждения устанавливают начальный и конечный момент стадии отверждения. Для целей настоящего изобретения начальный момент стадии отверждения определяют как момент достижения температуры отверждения. В некоторых вариантах на температурном профиле стадии отверждения имеется плато между начальной и конечной точками отверждения. В таких вариантах конечный момент стадии отверждения определяют как момент времени, когда нагревание прекращают или, по меньшей мере, уменьшают, например снижают нагрев и/или начинают следующую стадию охлаждения и, температура падает ниже температуры отверждения более чем примерно на 10 С и/или ниже нижнего предела интервала температур размягчения высокомолекулярного полиэтиленоксида, например ниже примерно 62 С. По достижении температуры отверждения, т.е. в начале стадии отверждения, в ходе стадии отверждения могут наблюдаться отклонения от температуры отверждения. Такие отклонения допустимы, если они не превышают примерно 10 С, предпочтительно примерно 6 С и более предпочтительно примерно 3 С. Например,- 11018311 когда надо поддерживать температуру отверждения по меньшей мере примерно 75 С, измеряемая температура может временно возрастать до значения примерно 85 С, предпочтительно примерно 81 С и более предпочтительно примерно 78 С, а также измеряемая температура может падать до значения примерно 65 С, предпочтительно примерно 69 С и более предпочтительно примерно 72 С. В случаях более заметного падения температуры и/или в случае, когда температура падает ниже нижнего предела интервала температур размягчения высокомолекулярного полиэтиленоксида, например ниже примерно 62 С, стадия отверждения останавливается, т.е. достигается конечный момент. Отверждение можно начать заново путем подъема температуры до температуры отверждения. В других вариантах температурный профиль стадии отверждения между начальным и конечным моментами стадии отверждения имеет форму параболы или треугольника. Это означает, что после начального момента, т.е. времени начала стадии отверждения, температура возрастает и достигает максимума, а затем снижается. В таких вариантах конечный момент определяют как момент времени, когда температура падает ниже температуры отверждения. В этом контексте следует отметить, что для характеризации температуры отверждения в зависимости от типа аппарата для отверждения, который далее будут называть устройством для отверждения,можно измерять разные температуры. В некоторых вариантах стадию отверждения можно проводить в сушильном шкафу. В таких вариантах измеряют температуру в сушильном шкафу. Когда стадия отверждения протекает в сушильном шкафу, то температуру отверждения определяют как заданную температуру внутри сушильного шкафа и началом стадии отверждения считают тот момент, когда температура достигает температуры отверждения. Конечным моментом стадии отверждения считают (1) тот момент времени, когда нагревание прекращают или, по меньшей мере, уменьшают, и затем температура в сушильном шкафу падает ниже температуры отверждения более чем примерно на 10 С и/или ниже нижнего предела интервала температур размягчения высокомолекулярного полиэтиленоксида, например ниже примерно 62 С, на плато температурного профиля или (2) момент времени, когда температура внутри сушильного шкафа падает ниже температуры отверждения на параболическом или треугольном температурном профиле. Предпочтительно, чтобы стадия отверждения начиналась тогда, когда температура внутри сушильного шкафа достигает температуры отверждения - по меньшей мере примерно 62 С, по меньшей мере примерно 68 С или по меньшей мере примерно 70 С, более предпочтительно по меньшей мере примерно 72 С или по меньшей мере примерно 75 С. В предпочтительных вариантах температурный профиль в ходе стадии отверждения имеет форму плато, причем температура отверждения, т.е. температура внутри сушильного шкафа, предпочтительно составляет по меньшей мере примерно 68 С, например, примерно 70 С или примерно 72 С, или примерно 73 С, или находится внутри интервала примерно 70-75 С, а время отверждения предпочтительно находится в интервале от примерно 30 мин до примерно 20 ч, более предпочтительно от примерно 30 мин до примерно 15 ч, или от примерно 30 мин до примерно 4 ч, или от примерно 30 мин до примерно 2 ч. Наиболее предпочтительно, чтобы время отверждения находилось в интервале от примерно 30 до примерно 90 мин. В некоторых других вариантах отверждение проводят в устройствах для отверждения, которые нагревают током воздуха с подачей нагретого воздуха (на входе) и его отводом, типа, например, дражировочного котла или кипящего слоя. Такие устройства для отверждения далее называют устройствами для отверждения с конвекцией. В таких устройствах для отверждения возможно замерять температуру входящего воздуха, т.е. температуру нагретого воздуха, входящего в устройство для отверждения с конвекцией, и/или температуру уходящего воздуха, т.е. температуру воздуха, покидающего устройство для отверждения с конвекцией. Можно также определять или, по меньшей мере, устанавливать температуру препаратов внутри устройства для отверждения с конвекцией во время стадии отверждения, например, с помощью инфракрасных приборов для измерения температуры, таких как ИК-пирометр, или с помощью датчиков температуры внутри устройства для отверждения, помещенных рядом с матричными формами препаратов с пролонгированным высвобождением. На основании этого при проведении стадии отверждения в устройстве для отверждения с конвекцией можно определить температуру отверждения и измерить время отверждения следующим образом. В одном варианте при измерении времени отверждения способом 1 температуру отверждения определяют как заданную температуру подаваемого воздуха и началом стадии отверждения считают момент времени, когда температура подаваемого воздуха достигает температуры отверждения. Концом стадии отверждения считают (1) момент времени, когда нагревание прекращают или, по меньшей мере, уменьшают, и температура подаваемого воздуха затем падает ниже температуры отверждения более чем примерно на 10 С и/или ниже нижнего предела интервала температур размягчения высокомолекулярного полиэтиленоксида, например ниже примерно 62 С, на плато температурного профиля или (2) момент времени, когда температура подаваемого воздуха падает ниже температуры отверждения на параболическом или треугольном температурном профиле. Предпочтительно, чтобы стадия отверждения начиналась по способу 1, когда температура подаваемого воздуха достигает температуры отверждения - по меньшей мере примерно 62 С, по меньшей мере примерно 68 С или по меньшей мере примерно 70 С,более предпочтительно по меньшей мере примерно 72 С или по меньшей мере примерно 75 С. В пред- 12018311 почтительном варианте температурный профиль во время стадии отверждения имеет вид плато, причем предпочтительно, чтобы температура отверждения, т.е. заданная температура подаваемого воздуха, составляла по меньшей мере примерно 72 С, например примерно 75 С, и предпочтительно, чтобы время отверждения, определенное по способу 1, находилось в интервале от примерно 15 мин до примерно 2 ч,например примерно от 30 мин до примерно 1 ч. В другом варианте, в котором время отверждения определяют по способу 2, температуру отверждения определяют как заданную температуру отходящего воздуха и началом стадии отверждения считают момент времени, когда температура отходящего воздуха достигает температуры отверждения. Концом стадии отверждения считают (1) момент времени, когда нагревание прекращают или, по меньшей мере,уменьшают, и затем температура отходящего воздуха падает ниже температуры отверждения более чем примерно на 10 С и/или ниже нижнего предела интервала температур размягчения высокомолекулярного полиэтиленоксида, например ниже примерно 62 С, на плато температурного профиля или (2) момент времени, когда температура отходящего воздуха падает ниже температуры отверждения на параболическом или треугольном температурном профиле. Предпочтительно, чтобы стадия отверждения начиналась по способу 2, когда температура отходящего воздуха достигает температуры отверждения по меньшей мере примерно 62 С, по меньшей мере примерно 68 С или по меньшей мере примерно 70 С, более предпочтительно по меньшей мере примерно 72 С или по меньшей мере примерно 75 С. В предпочтительных вариантах температурный профиль во время стадии отверждения имеет вид плато, причем предпочтительно, чтобы температура отверждения, т.е. заданная температура отходящего воздуха, составляла по меньшей мере примерно 68 С, по меньшей мере примерно 70 С или по меньшей мере примерно 72 С, например, заданная температура отходящего воздуха составляет примерно 68 С, примерно 70 С, примерно 72 С, примерно 75 С или примерно 78 С, причем предпочтительно, чтобы время отверждения, определенное по способу 2, находилось в интервале от примерно 1 мин до примерно 2 ч, предпочтительно от примерно 5 до примерно 90 мин, например, время отверждения составляет примерно 5 мин, примерно 10 мин, примерно 15 мин, примерно 30 мин, примерно 60 мин, примерно 70 мин, примерно 75 мин или примерно 90 мин. В более предпочтительном варианте время отверждения, которое определяют по способу 2, находится в интервале от примерно 15 мин до примерно 1 ч. В следующем варианте, в котором время отверждения определяют по способу 3, температуру отверждения определяют как заданную температуру матричных форм препарата с пролонгированным высвобождением и начало стадии отверждения определяют как момент времени, когда температура матричных форм препарата с пролонгированным высвобождением, которую можно определить с помощью,например, ИК-пирометра, достигает температуры отверждения. Конец стадии отверждения определяют как (1) момент времени, когда нагревание прекращают или, по меньшей мере, уменьшают, и затем температура матричных форм препарата с пролонгированным высвобождением падает ниже температуры отверждения более чем примерно на 10 С и/или ниже нижнего предела интервала температур размягчения высокомолекулярного полиэтиленоксида, например ниже примерно 62 С, на плато температурного профиля или (2) момент времени, когда температура матричных форм препарата с пролонгированным высвобождением падает ниже температуры отверждения на параболическом или треугольном температурном профиле. Предпочтительно, чтобы стадия отверждения начиналась по способу 3, когда температура матричных форм препарата с пролонгированным высвобождением достигает температуры отверждения, равной по меньшей мере примерно 62 С, по меньшей мере примерно 68 С или по меньшей мере примерно 70 С, более предпочтительно по меньшей мере примерно 72 С или по меньшей мере примерно 75 С. В еще одном варианте, в котором время отверждения определяют по способу 4, температуру отверждения определяют как заданную температуру, измеренную с помощью датчика температуры, например проволочной термопары, помещенной внутри устройства для отверждения рядом с матричными формами препарата с пролонгированным высвобождением; при этом начало стадии отверждения определяют как момент времени, когда температура, измеренная с помощью датчика температуры внутри устройства для отверждения рядом с матричными формами препарата с пролонгированным высвобождением, достигает температуры отверждения. Конец стадии отверждения определяют как (1) момент времени, когда нагревание прекращают или, по меньшей мере, уменьшают, и затем температура, измеренная с помощью датчика температуры, падает ниже температуры отверждения более чем примерно на 10 С и/или ниже нижнего предела интервала температур размягчения высокомолекулярного полиэтиленоксида, например ниже примерно 62 С, на плато температурного профиля или (2) момент времени, когда температура, измеренная с помощью датчика температуры, падает ниже температуры отверждения на параболическом или треугольном температурном профиле. Предпочтительно, чтобы стадия отверждения начиналась по способу 4, когда температура, измеренная с помощью датчика температуры внутри устройства для отверждения рядом с матричными формами препарата с пролонгированным высвобождением, достигает температуры отверждения по меньшей мере примерно 62 С, по меньшей мере примерно 68 С или по меньшей мере примерно 70 С, более предпочтительно по меньшей мере примерно 72 С или по меньшей мере примерно 75 С. В предпочтительном варианте температурный профиль во время стадии отверждения имеет форму плато, причем предпочтительно, чтобы температура отвержде- 13018311 ния, т.е. заданная температура, измеренная с помощью датчика температуры внутри устройства для отверждения рядом с матричными формами препарата с пролонгированным высвобождением, составляла по меньшей мере примерно 68 С, например примерно 70 С, и предпочтительно, чтобы время отверждения, которое определяют по способу 4, находилось в интервале от примерно 15 мин до примерно 2 ч,например, время отверждения составляет примерно 60 или примерно 90 мин. Если отверждение проводят в конвекционном устройстве для отверждения, время отверждения можно определить любым из способов (1, 2, 3 или 4. В предпочтительном варианте время отверждения определяют по способу 2. В некоторых вариантах температуру отверждения определяют как заданный температурный интервал, например температуру отверждения определяют как заданный интервал температур, например, подаваемого воздуха или заданный интервал температур отходящего воздуха. В таких вариантах начало стадии отверждения определяют как момент времени, при котором достигается нижний предел заданного интервала температур, а конец стадии отверждения определяют как момент времени, когда нагревание прекращают или, по меньшей мере, уменьшают, и температура последовательно падает ниже нижнего предела заданного интервала температур более чем примерно на 10 С и/или ниже нижнего предела интервала температур размягчения высокомолекулярного полиэтиленоксида, например ниже примерно 62 С. Время отверждения, т.е. период времени, в течение которого матричную форму препарата с пролонгированным высвобождением доводят до температуры отверждения, которую можно определить,например, способами 1, 2, 3 и 4, как описано выше, составляет по меньшей мере примерно 1 мин или по меньшей мере примерно 5 мин. Время отверждения может варьироваться от примерно 1 мин до примерно 24 ч, или от примерно 5 мин до примерно 20 ч, или от примерно 10 мин до примерно 15 ч, или от примерно 15 мин до примерно 10 ч, или от примерно 30 мин до примерно 5 ч в зависимости от конкретной композиции и от препарата и температуры отверждения. Параметр композиции, время отверждения и температуру отверждения выбирают такими, чтобы достичь указанной устойчивости к разрушению. Согласно некоторым вариантам время отверждения варьируют от примерно 15 до примерно 30 мин. Согласно другим вариантам, в которых температура отверждения составляет по меньшей мере примерно 60 С или по меньшей мере примерно 62 С, предпочтительно по меньшей мере примерно 68 С, по меньшей мере примерно 70 С, по меньшей мере примерно 72 С или по меньшей мере примерно 75 С или варьируется от примерно 62 до примерно 85 С или от примерно 65 до примерно 85 С, предпочтительно,чтобы время отверждения составляло по меньшей мере примерно 15 мин, по меньшей мере примерно 30 мин, по меньшей мере примерно 60 мин, по меньшей мере примерно 75 мин, по меньшей мере примерно 90 мин или примерно 120 мин. В предпочтительных вариантах, в которых температура отверждения составляет, например, по меньшей мере примерно 62 С, по меньшей мере примерно 68 С или по меньшей мере примерно 70 С, предпочтительно по меньшей мере примерно 72 С или по меньшей мере примерно 75 С или варьируется от примерно 62 до примерно 80 С, от примерно 65 до примерно 80 С, от примерно 68 до примерно 80 С, от примерно 70 до примерно 80 С или от примерно 72 до примерно 80 С, предпочтительно, чтобы время отверждения составляло по меньшей мере примерно 1 мин или по меньшей мере примерно 5 мин. Более предпочтительно, чтобы время отверждения составляло по меньшей мере примерно 10 мин, по меньшей мере примерно 15 мин или по меньшей мере примерно 30 мин. В некоторых таких вариантах время отверждения можно выбрать по возможности коротким, но достичь нужной устойчивости к разрушению. Например, предпочтительно, чтобы время отверждения не превышало примерно 5 ч, более предпочтительно оно не должно превышать примерно 3 ч и наиболее предпочтительно,чтобы оно не превышало примерно 2 ч. Предпочтительно, чтобы время отверждения находилось в интервале от примерно 1 мин до примерно 5 ч, от примерно 5 мин до примерно 3 ч, от примерно 15 мин до примерно 2 ч или от примерно 15 мин до примерно 1 ч. Любая комбинация температур отверждения и времен отверждения, как описано здесь, находится в объеме настоящего изобретения. В некоторых вариантах композицию только доводят до температуры отверждения, пока высокомолекулярный полиэтиленоксид, присутствующий в матричной форме препарата с пролонгированным высвобождением не достигнет температуры размягчения и/или, по меньшей мере, частично не расплавится. В некоторых таких вариантах время отверждения может быть менее примерно 5 мин, например, время отверждения может варьироваться от примерно 0 мин до примерно 3 ч, или от примерно 1 мин до примерно 2 ч, или от примерно 2 мин до примерно 1 ч. Возможно быстрое отверждение, если выбрать устройство для отверждения, в котором можно быстро нагреть высокомолекулярный полиэтиленоксид в матричной форме препарата с пролонгированным высвобождением, по меньшей мере, до температуры размягчения, так чтобы высокомолекулярный полиэтиленоксид, по меньшей мере, частично расплавился. Такими устройствами для отверждения являются, например, микроволновые печи, ультразвуковые устройства, аппараты со световым обучением, такие как аппараты с УФ-облучением, микроволновые(СВЧ)-печи или любые способы, известные специалистам в данной области. Специалистам известно, что размер матричной формы препарата с пролонгированным высвобождением может определять необходимое время отверждения и температуру отверждения, которые приводят к нужной устойчивости к разрушению. Не связывая себя какой-либо теорией, можно считать, что в случае крупной матричной формы препарата с пролонгированным высвобождением, такой как большая таблетка, необходим более длинный период отверждения для передачи тепла внутрь препарата, чем в случае соответствующего препарата меньшего размера. Более высокая температура повышает скорость теплопередачи и в результате уменьшает необходимое время отверждения. Стадию отверждения (c) можно проводить в сушильном шкафу. Лучше проводить стадию отверждения (c) в движущемся слое матричных форм препарата с пролонгированным высвобождением, как,например, в дражировочном котле. Дражировочный котел позволяет проводить стадию отверждения в периодическом режиме с последующей стадией нанесения покрытия без переноса лекарственных форм,например таблеток. Такой способ может включать стадии:(а) объединение (1) по меньшей мере одного полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000 и (2) по меньшей мере одного активного ингредиента с образованием композиции;(b) формование указанной композиции с образованием матричной формы препарата с пролонгированным высвобождением в форме таблетки прямым прессованием;(c) отверждение указанной таблетки нагреванием движущегося слоя таблеток до температуры примерно 62-90 С, предпочтительно примерно 70-90 С в течение по меньшей мере примерно 1 или по меньшей мере 5 мин, предпочтительно по меньшей мере примерно 30 мин в дражировочном котле и последовательным охлаждением слоя движущихся таблеток до температуры ниже примерно 50 С; и затем(d) нанесение покрытия на лекарственную форму в указанном дражировочном котле. В некоторых вариантах после стадии (d) нанесения покрытия на лекарственную форму можно проводить еще одну стадию отверждения. Дополнительную стадию отверждения можно проводить так же,как стадию (c). В некоторых таких вариантах предпочтительно, чтобы температура отверждения дополнительной стадии составляла по меньшей мере примерно 70 С, по меньшей мере примерно 72 С или по меньшей мере примерно 75 С, а время отверждения находилось в интервале от примерно 15 мин до примерно 1 ч, например примерно 30 мин. В некоторых вариантах в композицию добавляют антиоксидант, например BHT (бутилированный гидрокситолуол). В некоторых вариантах стадия отверждения (c) приводит к уменьшению плотности матричной формы препарата с пролонгированным высвобождением, так что плотность отвержденной матричной формы препарата с пролонгированным высвобождением оказывается ниже плотности матричной формы препарата с пролонгированным высвобождением до стадии отверждения (c). Предпочтительно, чтобы плотность отвержденной матричной формы препарата с пролонгированным высвобождением по сравнению с неотвержденной матричной формой препарата с пролонгированным высвобождением была ниже по меньшей мере примерно на 0,5%. Более предпочтительно, чтобы плотность отвержденной матричной формы препарата с пролонгированным высвобождением по сравнению с плотностью неотвержденной матричной формы препарата с пролонгированным высвобождением была ниже по меньшей мере примерно на 0,7%, по меньшей мере примерно 0,8%, по меньшей мере примерно 1,0%, по меньшей мере примерно 2,0% или по меньшей мере примерно 2,5%. Не связывая себя какой-либо теорией, можно полагать, что благодаря отсутствию повышенного давления на стадии (c) матричная форма препарата с пролонгированным высвобождением расширяется, что приводит к уменьшению плотности. Согласно следующему аспекту изобретения плотность матричной формы препарата с пролонгированным высвобождением в твердой лекарственной форме для перорального применения с пролонгированным высвобождением, предпочтительно содержащей оксикодон HCl в качестве активного ингредиента, равна или менее примерно 1,20 г/см 3. Предпочтительно, чтобы она была равна или меньше примерно 1,19 г/см 3, равна или меньше примерно 1,18 г/см 3 или равна или меньше примерно 1,17 г/см 3. Например,плотность матричной формы препарата с пролонгированным высвобождением находится в интервале примерно 1,10-1,20 г/см 3, примерно 1,11-1,20 г/см 3 или примерно 1,11-1,19 г/см 3. Предпочтительно, чтобы она находилась в интервале примерно 1,12-1,19 г/см 3 или примерно 1,13-1,19 г/см 3, более предпочтительно примерно 1,13-1,18 г/см 3. Плотность матричной формы препарата с пролонгированным высвобождением предпочтительно определять по принципу Архимеда с помощью жидкости с известной плотностью (0). Матричную форму препарата с пролонгированным высвобождением сначала взвешивают на воздухе и затем пропитывают жидкостью и взвешивают. Из этих двух взвешиваний плотность матричной формы препарата с пролонгированным высвобождениемможно определить по уравнению где- плотность матричной формы препарата с пролонгированным высвобождением; А - масса матричной формы препарата с пролонгированным высвобождением на воздухе; В - масса матричной формы препарата с пролонгированным высвобождением, смоченной жидкостью; 0 - плотность жидкости при данной температуре. Подходящей жидкостью с известной плотностью 0 является, например, гексан. Предпочтительно определять плотность матричной формы препарата с пролонгированным высвобождением с помощью весов Top-loading Mettler Toledo ModelAB 135-S/FACT, Serial1127430072 и комплекта для определения плотности 33360. Предпочтительно использовать гексан в качестве жидкости с известной плотностью 0. Приведенные в описании значения плотности соответствуют плотности матричной формы препарата с пролонгированным высвобождением при комнатной температуре. Предпочтительно относить плотность матричной формы препарата с пролонгированным высвобождением к плотности препарата без покрытия, например к плотности внутренней таблетки. В тех вариантах, где матричная форма препарата с пролонгированным высвобождением содержит покрытие, например, когда она уже прошла стадию нанесения покрытия (d) после стадии отверждения (c), предпочтительно определять плотность матричной формы препарата с пролонгированным высвобождением до стадии нанесения покрытия или удалять покрытие из матричной формы препарата с пролонгированным высвобождением с покрытием и затем определять плотность матричной формы препарата с пролонгированным высвобождением без покрытия. В описанных выше вариантах можно использовать высокомолекулярный полиэтиленоксид с молекулярной массой, по данным реологических измерений, примерно 2000000-5000000 или 20000008000000. В частности, можно использовать полиэтиленоксиды с молекулярной массой, по данным реологических измерений, примерно 2000000, 4000000, 7000000 или 8000000. Конкретно можно использовать полиэтиленоксиды с молекулярной массой, по данным реологических измерений, примерно 4000000. В тех вариантах, когда композиция содержит также по меньшей мере один низкомолекулярный полиэтиленоксид, можно использовать такие полиэтиленоксиды с примерной молекулярной массой, по данным реологических измерений, менее 1000000, например полиэтиленоксиды с молекулярной массой,по данным реологических измерений, примерно 100000-900000. Добавку таких низкомолекулярных полиэтиленоксидов можно использовать для конкретного регулирования скорости высвобождения, например для увеличения скорости высвобождения препарата или для замедления этого процесса в некоторых специальных целях. В таких вариантах можно использовать по меньшей мере один полиэтиленоксид с молекулярной массой примерно 100000, по данным реологических измерений, . В некоторых таких вариантах композиция содержит по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000 и по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, менее 1000000, причем композиция содержит по меньшей мере примерно 10 мас.% или по меньшей мере примерно 20 мас.% полиэтиленоксида с молекулярной массой, по данным реологических измерений,менее примерно 1000000. В некоторых таких вариантах температура отверждения составляет менее примерно 80 С или даже менее примерно 77 С. В некоторых вариантах общее содержание полиэтиленоксида в композиции составляет по меньшей мере примерно 80 мас.%. Не связывая себя какой-либо теорией, можно считать, что высокие концентрации полиэтиленоксида повышают устойчивость к разрушению, как описано выше, т.е. увеличивают прочность при разрушении, и устойчивость к экстракции спиртом. Согласно некоторым таким вариантам активный ингредиент представляет собой оксикодон гидрохлорид и композиция содержит более примерно 5 мас.% оксикодон гидрохлорида. В некоторых таких вариантах содержание в композиции по меньшей мере одного полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000 составляет по меньшей мере примерно 80 мас.%. В некоторых вариантах содержание в композиции по меньшей мере одного полиэтиленоксида с молекулярной массой, по данным реологических измерений,по меньшей мере примерно 1000000 составляет по меньшей мере примерно 85 мас.% или по меньшей мере примерно 90 мас.%. В таких вариантах можно использовать полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 4000000 или по меньшей мере 7000000. В некоторых таких вариантах активный ингредиент представляет собой оксикодон гидрохлорид или гидроморфон гидрохлорид, хотя согласно этому аспекту изобретения можно использовать и другие активные ингредиенты и композиция будет содержать более примерно 5 мас.% оксикодон гидрохлорида или гидроморфон гидрохлорида. В некоторых вариантах, когда количество лекарства в композиции составляет по меньшей мере примерно 20 мас.%, полиэтиленоксид может составлять всего примерно 75 мас.%. В другом варианте,когда количество лекарства в композиции находится в интервале примерно 25-35 мас.%, полиэтиленоксид может составлять примерно 65-75 мас.%. Например, в вариантах, когда количество лекарства в композиции составляет примерно 32 мас.%, полиэтиленоксид может составлять примерно 67 мас.%. В некоторых вариантах изобретения во время или после осуществления способа отверждения/стадии отверждения добавляют стеарат магния, с тем чтобы избежать слипания таблеток. В некоторых таких вариантах стеарат магния добавляют в конце процесса отверждения/стадии отверждения перед охлаждением таблеток или во время охлаждения таблеток. Другими реагентами против слипания могли бы быть тальк, оксид кремния, коллоидный диоксид кремния, стеарат кальция, воск карнаубы,длинноцепные жирные спирты и воски, такие как стеариновая кислота и стеариловый спирт, минеральное масло, парафин, микрокристаллическая целлюлоза, глицерин, пропиленгликоль и полипропиленгликоль. Наряду с этим или альтернативно нанесение покрытия можно начинать при высокой температуре. В некоторых вариантах, когда стадию отверждения (c) проводят в дражировочном котле, слипания таблеток можно избежать или слипшиеся таблетки можно разделить путем увеличения скорости вращения котла во время или после стадии отверждения, причем в последнем случае, например, до или во время охлаждения таблеток. Скорость вращения котла увеличивают до такой, при которой все таблетки разделяются и не слипаются. В некоторых вариантах изобретения начальное пленочное покрытие или часть пленочного покрытия наносят до проведения стадии отверждения (c). Это пленочное покрытие образует "оболочку" для матричных форм препаратов с пролонгированным высвобождением или таблеток, которая функционирует как антиадгезив, т.е. помогает избежать слипания препаратов или таблеток. В некоторых таких вариантах пленочное покрытие, которое наносят до стадии отверждения, представляет собой пленку Opadry. После стадии отверждения (c) можно провести следующую стадию нанесения пленки. Настоящее изобретение охватывает также любые твердые лекарственные формы для перорального применения с пролонгированным высвобождением, получаемые по любому способу, описанному выше. Независимо настоящее изобретение относится также к твердым лекарственным формам для перорального применения с пролонгированным высвобождением. В некоторых вариантах изобретение относится к твердым лекарственным формам для перорального применения с пролонгированным высвобождением, содержащим матричные формы препаратов с пролонгированным высвобождением, включающие активный ингредиент, в форме таблеток или множества частиц, причем таблетку или множество отдельных частиц можно, по меньшей мере, сплющить без разрушения, так что толщина таблетки или отдельных частиц из множества после сплющивания составляет не более чем примерно 60% от толщины таблетки или отдельной частицы из множества частиц до сплющивания, и причем указанная сплющенная таблетка или множество сплющенных частиц обеспечивают такую скорость растворения in vitro при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, что количество активного ингредиента в процентах, высвободившегося за 0,5 ч, или за 0,5 и 0,75 ч, или за 0,5,0,75 и 1 ч, или за 0,5, 0,75, 1 и 1,5 ч, или за 0,5, 0,75, 1, 1,5 и 2 ч растворения, отличается не более чем на 20% пунктов в каждый указанный момент времени от соответствующей скорости растворения in vitro несплющенной эталонной таблетки или множества эталонных частиц. В некоторых таких вариантах таблетку или множество отдельных частиц можно, по меньшей мере,сплющить без разрушения, причем толщина таблетки или отдельных частиц из множества после сплющивания составляет не более чем примерно 50%, или не более чем примерно 40%, или не более чем примерно 30%, или не более чем примерно 20%, или не более чем примерно 16% от толщины таблетки или отдельной частицы из множества частиц до сплющивания, и причем указанная сплющенная таблетка или множество сплющенных частиц обеспечивают такую скорость растворения in vitro при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, что количество активного ингредиента в процентах, высвободившегося за 0,5 ч, или за 0,5 и 0,75 ч, или за 0,5, 0,75 и 1 ч, или за 0,5, 0,75, 1 и 1,5 ч, или за 0,5, 0,75, 1, 1,5 и 2 ч растворения, отличается не более чем на 20% пунктов или не более чем на 15% пунктов в каждый указанный момент времени от соответствующей скорости растворения in vitro несплющенных эталонной таблетки или множества эталонных частиц. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, включающую активный ингредиент, в форме таблетки или множества частиц, причем таблетку или множество отдельных частиц можно, по меньшей мере, сплющить без разрушения, причем толщина таблетки или отдельных частиц из множества после сплющивания составляет не более чем примерно 60% от толщины таблетки или отдельной частицы из множества частиц до сплющивания, и причем указанная сплющенная таблетка или множество сплющенных частиц и несплющенная эталонная таблетка или множество эталонных частиц обеспечивают такую скорость растворения in vitro при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, при которой количество активного ингредиента, высвободившегося за 0,5 ч, составляет примерно 5-40 мас.%. В некоторых таких вариантах таблетку или множество отдельных частиц можно, по меньшей мере,сплющить без разрушения, причем толщина таблетки или отдельных частиц из множества после сплющивания соответствует не более чем примерно 50%, или не более чем примерно 40%, или не более чем примерно 30%, или не более чем примерно 20%, или не более чем примерно 16% от толщины таблетки или отдельной частицы из множества частиц до сплющивания, и причем указанная сплющенная таблетка или множество сплющенных частиц и несплющенная эталонная таблетка или множество эталонных частиц обеспечивают такую скорость растворения in vitro при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, при которой количество активного ингредиента, высвободившегося за 0,5 ч, составляет примерно 5-40 мас.%,или примерно 5-30 мас.%, или примерно 5-20 мас.%, или примерно 10-18 мас.%. В некоторых вариантах изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, включающую активный ингредиент, в форме таблетки или множества частиц, причем таблетку или множество отдельных частиц можно, по меньшей мере, сплющить без разрушения, причем толщина таблетки или отдельных частиц из множества частиц после сплющивания соответствует не более чем примерно 60% толщины таблетки или отдельной частицы из множества частиц до сплющивания, и причем указанная сплющенная или несплющенная таблетка или множество сплющенных или несплющенных частиц обеспечивают такую скорость растворения in vitro при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF) и содержащего 40% этанол, при 37 С, при которой количество активного ингредиента в процентах, высвободившегося за 0,5 ч, или за 0,5 и 0,75 ч, или за 0,5, 0,75 и 1 ч, или за 0,5, 0,75, 1 и 1,5 ч,или за 0,5, 0,75, 1, 1,5 и 2 ч растворения, отличается не более чем примерно на 20% пунктов в каждый момент времени от соответствующей скорости растворения in vitro, определенной на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, без этанола, сплющенной или несплющенной эталонной таблетки или множества эталонных сплющенных или несплющенных частиц. В некоторых вариантах таблетку или множество отдельных частиц можно, по меньшей мере,сплющить без разрушения, причем толщина таблетки или отдельных частиц из множества после сплющивания соответствует не более чем примерно 60%, или не более чем примерно 50%, или не более чем примерно 40%, или не более чем примерно 30%, или не более чем примерно 20%, или не более чем примерно 16% от толщины таблетки или частиц из множества отдельных частиц до сплющивания, и причем указанная сплющенная и несплющенная таблетка или частица из множества частиц обеспечивают такую скорость растворения in vitro при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF) и содержащего 40% этанол, при 37 С,что количество активного ингредиента в процентах, высвободившегося за 0,5 ч, или за 0,5 и 0,75 ч, или за 0,5, 0,75 и 1 ч, или за 0,5, 0,75, 1 и 1,5 ч, или за 0,5, 0,75, 1, 1,5 и 2 ч растворения, отличается не более чем примерно на 20% пунктов или не более чем примерно на 15% пунктов в каждый указанный момент времени от соответствующей скорости растворения in vitro, определенной на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, без этанола, сплющенной или несплющенной эталонной таблетки или множества эталонных частиц соответственно. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, включающую активный ингредиент, в форме таблетки или множества частиц, причем таблетку или множество отдельных частиц можно, по меньшей мере, сплющить без разрушения, причем толщина таблетки или отдельных частиц из множества частиц после сплющивания соответствует не более чем примерно 60% от толщины таблетки или отдельной частицы из множества частиц до сплющивания, и причем указанная сплющенная или несплющенная таблетка или множество сплющенных и несплющенных частиц обеспечивают такую скорость растворения in vitro при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF) и содержащего 40 или 0% этанола, при 37 С, при которой за 0,5 ч высвобождается примерно 5-40 мас.% активного ингредиента. В некоторых вариантах таблетку или множество отдельных частиц можно, по меньшей мере,сплющить без разрушения, причем толщина таблетки или отдельных частиц из множества после сплющивания составляет не более чем примерно 50%, или не более чем примерно 40%, или не более чем примерно 30%, или не более чем примерно 20%, или не более чем примерно 16% от толщины таблетки или частиц из множества отдельных частиц до сплющивания, и причем указанная сплющенная или несплющенная таблетка или множество сплющенных или несплющенных частиц обеспечивают такую скорость растворения in vitro при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF) и содержащего 40 или 0% этанола, при 37 С,при которой за 0,5 ч высвобождается примерно 5-40 мас.% активного ингредиента, или примерно 5-30 мас.%, или примерно 5-20 мас.%, или примерно 10-18 мас.% активного ингредиента. Такие лекарственные формы можно изготовить, как описано выше. В некоторых вариантах изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, включающую композицию, состоящую из:(1) по меньшей мере одного полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000;(2) по меньшей мере одного активного ингредиента, который предпочтительно выбирать из опиоидных анальгетиков; причем композиция содержит по меньшей мере примерно 80 мас.% полиэтиленоксида. Композиция может также содержать по меньшей мере примерно 85-90 мас.% полиэтиленоксида. Согласно некоторым вариантам, в которых композиция содержит по меньшей мере примерно 80 мас.% полиэтиленоксида,активный ингредиент представляет собой оксикодон гидрохлорид или гидроморфон гидрохлорид и композиция содержит более примерно 5 мас.% оксикодон гидрохлорида или гидроморфон гидрохлорида. В некоторых из таких вариантов композиция содержит по меньшей мере примерно 80 мас.% полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением включает композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000;(2) по меньшей мере 10 мг оксикодон гидрохлорида; и в которой композиция содержит по меньшей мере примерно 85 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением включает композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000;(2) по меньшей мере 15-20 мг оксикодон гидрохлорида; причем композиция содержит по меньшей мере примерно 80 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением включает композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000;(2) по меньшей мере 40 мг оксикодон гидрохлорида; причем композиция содержит по меньшей мере примерно 65 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением включает композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000;(2) по меньшей мере 60-80 мг оксикодон гидрохлорида; причем композиция содержит по меньшей мере примерно 60 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением включает композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000;(2) по меньшей мере 8 мг гидроморфон гидрохлорида; причем композиция содержит по меньшей мере примерно 94 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением включает композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000;(2) по меньшей мере 12 мг гидроморфон гидрохлорида; причем композиция содержит по меньшей мере примерно 92 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением включает композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000;(2) по меньшей мере 32 мг гидроморфон гидрохлорида; эта композиция содержит по меньшей мере примерно 90 мас.% полиэтиленоксида. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением включает композицию, содержащую:(1) по меньшей мере один активный ингредиент, который предпочтительно выбирать из опиоидных анальгетиков;(2) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000;(3) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, менее примерно 1000000. В некоторых таких вариантах композиция содержит по меньшей мере примерно 80 мас.% полиэтиленоксида. Композиция может также содержать по меньшей мере примерно 85-90 мас.% полиэтиленоксида. Согласно некоторым таким вариантам, в которых композиция содержит по меньшей мере примерно 80 мас.% полиэтиленоксида, активный ингредиент представляет собой оксикодон гидрохлорид или гидроморфон гидрохлорид и композиция содержит более примерно 5 мас.% оксикодон гидрохлорида или гидроморфон гидрохлорида. Композиция может также содержать 15-30 мас.% полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000; 65-80 мас.% полиэтиленоксида с молекулярной массой, по данным реологических измерений, менее 1000000 или композиция может содержать по меньшей мере примерно 20 мас.%, или по меньшей мере примерно 30 мас.%, или по меньшей мере примерно 50 мас.% полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере 1000000. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением включает композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере 800000 или по меньшей мере 900000;(2) по меньшей мере один активный ингредиент, который выбирают из опиоидных анальгетиков; и эта композиция содержит по меньшей мере примерно 80 мас.% полиэтиленоксида. В некоторых вариантах изобретения матрица с пролонгированным высвобождением имеет плотность, равную или меньше примерно 1,20 г/см 3. В некоторых вариантах плотность матричной формы с пролонгированным высвобождением равна или меньше примерно 1,19 г/см 3, предпочтительно равна или меньше примерно 1,18 г/см 3, или равна или меньше примерно 1,17 г/см 3. Например, плотность матричной формы с пролонгированным высвобождением находится в интервале примерно 1,10-1,20 г/см 3, примерно 1,11-1,20 г/см 3 или примерно 1,11-1,19 г/см 3. Предпочтительно, чтобы она была в интервале примерно 1,12-1,19 г/см 3 или примерно 1,13-1,19 г/см 3, более предпочтительно примерно 1,13-1,18 г/см 3. Предпочтительно определять плотность по принципу Архимеда, как описано выше. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением включает композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000;(2) по меньшей мере один активный ингредиент, и причем матричная форма препарата с пролонгированным высвобождением в тесте на вдавливание характеризуется разрушающим усилием по меньшей мере примерно 110 Н. В некоторых вариантах изобретения матричная форма препарата с пролонгированным высвобождением характеризуется разрушающим усилием по меньшей мере примерно 110 Н, предпочтительно по меньшей мере примерно 120 Н, по меньшей мере примерно 130 Н или по меньшей мере примерно 140 Н,более предпочтительно по меньшей мере примерно 150 Н, по меньшей мере примерно 160 Н или по меньшей мере примерно 170 Н, наиболее предпочтительно по меньшей мере примерно 180 Н, по меньшей мере примерно 190 Н или по меньшей мере примерно 200 Н. В некоторых вариантах настоящее изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением включает композицию, содержащую:(1) по меньшей мере один полиэтиленоксид с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000;(2) по меньшей мере один активный ингредиент, и причем матричная форма препарата с пролонгированным высвобождением в тесте на вдавливание характеризуется "глубиной проникновения до растрескивания" по меньшей мере примерно 1,0 мм. В некоторых вариантах изобретения матричная форма препарата с пролонгированным высвобождением в тесте на вдавливание характеризуется "глубиной проникновения до растрескивания" по меньшей мере примерно 1,0 мм или по меньшей мере примерно 1,2 мм, предпочтительно по меньшей мере примерно 1,4 мм, по меньшей мере примерно 1,5 мм или по меньшей мере примерно 1,6 мм, более предпочтительно по меньшей мере примерно 1,8 мм, по меньшей мере примерно 1,9 мм или по меньшей мере примерно 2,0 мм, наиболее предпочтительно по меньшей мере примерно 2,2 мм, по меньшей мере примерно 2,4 мм или по меньшей мере примерно 2,6 мм. В некоторых таких вариантах изобретения матричная форма препарата с пролонгированным высвобождением характеризуется разрушающим усилием по меньшей мере примерно 110 Н, предпочтительно по меньшей мере примерно 120 Н, по меньшей мере примерно 130 Н или по меньшей мере примерно 140 Н, более предпочтительно по меньшей мере примерно 150 Н, по меньшей мере примерно 160 Н или по меньшей мере примерно 170 Н, наиболее предпочтительно по меньшей мере примерно 180 Н, по меньшей мере примерно 190 Н или по меньшей мере примерно 200 Н и/или "глубиной проникновения до растрескивания" по меньшей мере примерно 1,0 мм или по меньшей мере примерно 1,2 мм, предпочтительно по меньшей мере примерно 1,4 мм, по меньшей мере примерно 1,5 мм или по меньшей мере примерно 1,6 мм, более предпочтительно по меньшей мере примерно 1,8 мм, по меньшей мере примерно 1,9 мм или по меньшей мере примерно 2,0 мм, наиболее предпочтительно по меньшей мере примерно 2,2 мм, по меньшей мере примерно 2,4 мм или по меньшей мере примерно 2,6 мм. Комбинация любых указанных величин разрушающего усилия и "глубины проникновения до растрескивания" входит в объем настоящего изобретения. В некоторых вариантах матричная форма препарата с пролонгированным высвобождением в тесте на вдавливание сохраняет свою форму без разрушения при нагрузке примерно 0,06 Дж или по меньшей мере примерно 0,08 Дж, предпочтительно по меньшей мере примерно 0,09 Дж, по меньшей мере примерно 0,11 Дж или по меньшей мере примерно 0,13 Дж, более предпочтительно по меньшей мере примерно 0,15 Дж, по меньшей мере примерно 0,17 Дж или по меньшей мере примерно 0,19 Дж, наиболее предпочтительно по меньшей мере примерно 0,21 Дж, по меньшей мере примерно 0,23 Дж или по меньшей мере примерно 0,25 Дж. Параметры "разрушающее усилие", "глубина проникновения до растрескивания" и "нагрузка" определяют в тесте на вдавливание, как описано выше, с помощью текстурного анализатора типа TA-ХТ 2Texture Analyzer (Texture Technologies Corp., 18 Fairview Road, Scarsdale, NY 10583). Разрушающее усилие и/или "глубину проникновения до растрескивания" можно определять для матричной формы препарата с пролонгированным высвобождением без покрытия или с покрытием. Предпочтительно определять разрушающее усилие и/или "глубину проникновения до растрескивания" для матричной формы препарата с пролонгированным высвобождением без покрытия. Не обращаясь к теории, можно полагать, что покрытие, нанесенное на стадии (d) способа производства твердой лекарственной формы для перорального применения с пролонгированным высвобождением, как описано выше, не вносит заметного вклада в величины разрушающего усилия и/или "глубины проникновения до растрескивания". Поэтому нельзя ожидать, что разрушающее усилие и/или "глубина проникновения до растрескивания", определенные для конкретной матричной формы препарата с пролонгированным высвобождением с покрытием, будут заметно отличаться от значений, найденных для соответствующей матричной формы препарата с пролонгированным высвобождением без покрытия. В некоторых вариантах матричная форма препарата с пролонгированным высвобождением имеет вид таблетки или множества частиц, и таблетку или множество отдельных частиц можно, по меньшей мере, сплющить без разрушения, причем толщина таблетки или множества отдельных частиц после сплющивания составляет не более чем примерно 60% от толщины таблетки или множества отдельных частиц до сплющивания. Предпочтительно, чтобы таблетку или множество отдельных частиц можно было, по меньшей мере, сплющить без разрушения и толщина таблетки или множества отдельных частиц после сплющивания составляла не более чем примерно 50%, или не более чем примерно 40%, или не более чем примерно 30%, или не более чем примерно 20%, или не более чем примерно 16% от толщины таблетки или множества отдельных частиц до сплющивания. Предпочтительно сплющивать таблетки или множество отдельных частиц на настольном прессе или молотком, как описано выше. В некоторых таких вариантах матричная форма препарата с пролонгированным высвобождением имеет форму таблетки или множества частиц, и эту таблетку или множество отдельных частиц можно,по меньшей мере, сплющить без разрушения, причем толщина таблетки или отдельных частиц из множества после сплющивания составляет не более чем примерно 60% от толщины таблетки или отдельной частицы из множества частиц до сплющивания, и причем указанная сплющенная таблетка или множество сплющенных частиц обеспечивают скорость растворения in vitro, определенную на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, отличающуюся тем, что количество активного ингредиента в процентах, высвободившегося за 0,5 ч, или за 0,5 и 0,75 ч, или за 0,5, 0,75 и 1 ч, или за 0,5, 0,75, 1 и 1,5 ч, или за 0,5, 0,75, 1, 1,5 и 2 ч растворения, отличается не более чем на 20% пунктов в каждый указанный момент времени от соответствующей скорости растворения in vitro несплющенных эталонной таблетки или множества эталонных частиц. Предпочтительно, чтобы таблетку или множество отдельных частиц можно было, по меньшей мере, сплющить без разрушения, и толщина таблетки или отдельных частиц из множества после сплющивания составляла не более чем примерно 50%, или не более чем примерно 40%, или не более чем примерно 30%, или не более чем примерно 20%, или не более чем примерно 16% от толщины таблетки или отдельной частицы из множества частиц до сплющивания, и причем скорость растворения in vitro указанной сплющенной таблетки или множества сплющенных частиц при определении на Аппарате USP 1(с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF),при 37 С, такова, что количество активного ингредиента в процентах, высвободившегося за 0,5 ч, или за 0,5 и 0,75 ч, или за 0,5, 0,75 и 1 ч, или за 0,5, 0,75, 1 и 1,5 ч, или за 0,5, 0,75, 1, 1,5 и 2 ч растворения, отличается не более чем на 20% пунктов или не более чем на 15% пунктов в каждый указанный момент времени от соответствующей скорости растворения in vitro несплющенной эталонной таблетки или множества эталонных частиц. В некоторых вариантах изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, содержащей матричную форму препарата с пролонгированным высвобождением, причем матричная форма препарата с пролонгированным высвобождением имеет форму таблеток или множества частиц, и таблетку или множество отдельных частиц можно, по меньшей мере, сплющить без разрушения, так что толщина таблетки или отдельных частиц из множества после сплющивания составляет не более чем примерно 60% от толщины таблетки или отдельной частицы из множества частиц до сплющивания, причем скорость растворения in vitro указанной сплющенной или несплющенной таблетки или множества сплющенных и несплющенных частиц при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF) и содержащего 40% этанол, при 37 С, такова, что количество активного ингредиента в процентах, высвободившегося за 0,5 ч, или за 0,5 и 0,75 ч, или за 0,5, 0,75 и 1 ч, или за 0,5, 0,75, 1 и 1,5 ч,или за 0,5, 0,75, 1, 1,5 и 2 ч растворения, отличается не более чем на 20% пунктов в каждый указанный момент времени от соответствующей скорости растворения in vitro, определенной на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF) без этанола, при 37 С, для сплющенной и несплющенной эталонной таблетки или множества несплющенных эталонных частиц соответственно. Предпочтительно, чтобы таблетку или множество отдельных частиц можно было, по меньшей мере, сплющить без разрушения и толщина таблетки или множества отдельных частиц после сплющивания составляет не более чем примерно 60%, не более чем примерно 50%, или не более чем примерно 40%, или не более чем примерно 30%, или не более чем примерно 20%, или не более чем примерно 16% от толщины таблетки или множества отдельных частиц до сплющивания, причем скорость растворения in vitro указанной сплющенной или несплющенной таблетки или множества отдельных частиц при определении на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF) и содержащего 40% этанол, при 37 С, такова, что количество активного ингредиента в процентах, высвободившегося за 0,5 ч, или за 0,5 и 0,75 ч, или за 0,5, 0,75 и 1 ч, или за 0,5, 0,75, 1 и 1,5 ч, или за 0,5, 0,75, 1, 1,5 и 2 ч растворения, отличается не более чем на 20% пунктов или не более чем на 15% пунктов в каждый указанный момент времени от соответствующей скорости растворения in vitro, определенной на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, без этанола, для сплющенной и несплющенной эталонной таблетки или множества эталонных частиц соответственно. В некоторых таких вариантах матричная форма препарата с пролонгированным высвобождением в тесте на твердость таблетки не разрушается при максимальном усилии примерно 196 Н или примерно 439 Н. Предпочтительно проводить тест на твердость таблетки для определения прочности к разрушению матричной формы препарата с пролонгированным высвобождением в аппарате Schleuniger, как описано выше. Например, прочность к разрушению определяют в аппарате Schleuniger 2E/106, прилагая усилие максимум примерно 196 Н, или в аппарате Schleuniger Model 6D, прилагая усилие максимум примерно 439 Н. Было установлено также, что препарат настоящего изобретения устойчив при хранении, причем матричная форма препарата с пролонгированным высвобождением после хранения при 25 С и 60% относительной влажности (отн. влажн.) или 40 С и 75% относительной влажности (отн. влажн.) в течение по меньшей мере 1 месяца, более предпочтительно в течение по меньшей мере 2 месяцев, в течение по меньшей мере 3 месяцев или в течение по меньшей мере примерно 6 месяцев растворяется со скоростью,определенной на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, при которой количество активного ингредиента, высвободившегося за 1 ч, или за 1 и 2 ч, или за 1 и 4 ч, или за 1, 2 и 4 ч, или за 1, 4 и 12 ч, или за 1, 2, 4 и 8 ч, или за 1,2, 4, 8 и 12 ч растворения, отличается не более чем примерно на 15% пунктов, предпочтительно не более чем примерно на 12% пунктов или не более чем примерно на 10% пунктов, более предпочтительно не более чем примерно на 8% пунктов или не более чем примерно на 6% пунктов, наиболее предпочтительно не более чем примерно на 5% пунктов в каждый момент времени от соответствующей скорости растворения in vitro эталонного препарата до хранения. Предпочтительно хранить матричную форму препарата с пролонгированным высвобождением в мерных бутылках, например в 100-мерных бутылках. Любая комбинация указанных выше времен хранения, моментов растворения и пределов отклонений входит в объем настоящего изобретения. Согласно следующему аспекту устойчивости матричная форма препарата с пролонгированным высвобождением после хранения при 25 С и 60% относительной влажности (отн. влажн.) или 40 С и 75% относительной влажности (отн. влажн.) в течение по меньшей мере 1 месяца, более предпочтительно в течение по меньшей мере 2 месяцев, в течение по меньшей мере 3 месяцев или по меньшей мере в течение примерно 6 месяцев содержит количество по меньшей мере одного активного ингредиента, которое отличается не более чем примерно на 10% пунктов, предпочтительно не более чем примерно на 8% пунктов или не более чем примерно на 6% пунктов, более предпочтительно не более чем примерно на 4% пунктов или не более чем примерно на 3% пунктов от указанного на этикетке количества соответствующего активного ингредиента в мас.% в матричной форме эталонного препарата до хранения. Предпочтительно хранить матричную форму препарата с пролонгированным высвобождением в мерных бутылках, например в 100-мерных бутылках. Любая комбинация указанных выше времен хранения и пределов отклонений входит в объем настоящего изобретения. Согласно некоторым вариантам активный ингредиент представляет собой оксикодон гидрохлорид. Предпочтительно определять количество по меньшей мере одного активного ингредиента в мас.% относительно указанного на этикетке количества активного ингредиента в матричной форме препарата с пролонгированным высвобождением путем экстракции по меньшей мере одного активного ингредиента из матричной формы препарата с пролонгированным высвобождением и последующего анализа методом ВЭЖХ. В некоторых вариантах, в которых по меньшей мере один активный ингредиент представляет собой оксикодон гидрохлорид, предпочтительно определять количество оксикодон гидрохлорида в мас.% относительно указанного на этикетке количества оксикодон гидрохлорида в матричной форме препарата с пролонгированным высвобождением путем экстракции оксикодон гидрохлорида из матричной формы препарата с пролонгированным высвобождением смесью 1:2 ацетонитрила и модельного желудочного сока, не содержащего ферментов (SGF), при постоянном перемешивании магнитной мешалкой до полного диспергирования матричной формы препарата с пролонгированным высвобождением или в течение ночи и последующего анализа методом ВЭЖХ, предпочтительно обращенно-фазовой ВЭЖХ. В некоторых таких вариантах, в которых матричная форма препарата с пролонгированным высвобождением находится в форме таблеток, предпочтительно определять количество оксикодон гидрохлорида в мас.% относительно указанного на этикетке количества оксикодон гидрохлорида в таблетках путем экстракции оксикодон гидрохлорида из двух серий по десять таблеток каждая с помощью 900 мл смеси 1:2 ацетонитрила и модельного желудочного сока, не содержащего ферментов (SGF), при постоянном перемешивании с помощью магнитной мешалки до полного диспергирования таблеток или в течение ночи и последующего анализа методом ВЭЖХ, предпочтительно обращенно-фазовой ВЭЖХ. Предпочтительно, чтобы количественные результаты анализа были средними значениями из двух измерений. В некоторых вариантах изобретение относится к твердой лекарственной форме для перорального применения с пролонгированным высвобождением, скорость растворения которой, определенная на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, такова, что при этом высвобождается 12,5-55 мас.% активного ингредиента за 1 ч, примерно 25-65 мас.% активного ингредиента за 2 ч, примерно 45-85 мас.% активного ингредиента за 4 ч и примерно 55-95 мас.% активного ингредиента за 6 ч и, необязательно, примерно 75-100 мас.% активного ингредиента за 8 ч. Предпочтительно, чтобы скорость растворения лекарственной формы, определенная на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, была такова, что 15-45 мас.% активного ингредиента высвобождается за 1 ч, примерно 30-60 мас.% активного ингредиента за 2 ч, примерно 50-80 мас.% активного ингредиента за 4 ч и примерно 60-90 мас.% активного ингредиента за 6 ч и, необязательно, примерно 80-100 мас.% активного ингредиента за 8 ч. Более предпочтительно, чтобы лекарственная форма растворялась с такой скоростью, определенной на Аппарате USP 1 (с корзиной) при 100 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, что 17,5-35 мас.% активного ингредиента высвобождается за 1 ч, примерно 35-55 мас.% активного ингредиента за 2 ч, примерно 55-75 мас.% активного ингредиента за 4 ч и примерно 65-85 мас.% активного ингредиента за 6 ч и, необязательно, примерно 85-100 мас.% активного ингредиента за 8 ч. В некоторых таких вариантах активный ингредиент представляет собой оксикодон гидрохлорид или гидроморфон гидрохлорид. Такие лекарственные формы можно изготовить описанным выше способом. В описанных выше вариантах таблетку можно формовать прямым прессованием композиции и от- 23018311 верждать, по меньшей мере, путем нагревания указанной таблетки до температуры по меньшей мере примерно 60 С, по меньшей мере примерно 62 С, по меньшей мере примерно 68 С, по меньшей мере примерно 70 С, по меньшей мере примерно 72 С или по меньшей мере примерно 75 С в течение примерно 1 мин, по меньшей мере примерно 5 мин или по меньшей мере примерно 15 мин. В некоторых вариантах указанную выше таблетку можно покрыть сверху слоем порошка полиэтиленоксида, нанося на отвержденную или неотвержденную таблетку слой порошка полиэтиленоксида,окружающий ядро, и отверждая таблетку со слоем порошка, как описано выше. Такой наружный слой полиэтиленоксида требует некоторого времени для высвобождения активного ингредиента и/или обеспечивает более низкую общую скорость высвобождения. В некоторых вариантах изобретения получают двухслойную и многослойную слипшуюся таблетку,в которой по меньшей мере один из слоев содержит описанную выше матричную форму препарата с пролонгированным высвобождением, и по меньшей мере один из остальных слоев содержит препарат немедленного высвобождения активного ингредиента, содержащийся в препарате с пролонгированным высвобождением, или второй активный ингредиент. В некоторых таких вариантах таблетка представляет собой двухслойную таблетку со слоем препарата с пролонгированным высвобождением, как здесь описано, и слоем препарата с немедленным высвобождением. В некоторых таких вариантах, в частности в двухслойных таблетках, опиоидные анальгетики находятся в слое с пролонгированным высвобождением, а неопиоидные анальгетики содержатся в слое с немедленным высвобождением. Неопиоидные анальгетики могут быть нестероидными противовоспалительными препаратами, а также неопиоидными анальгетиками, такими как ацетаминофен. Ацетаминофен можно, например, использовать в комбинации с гидрокодоном как опиоидным анальгетиком. Такие таблетки можно изготовить по специальной методике прессования таблеток, позволяющей прессовать по меньшей мере две композиции в таблетки, состоящие по меньшей мере из двух соединенных разных слоев, каждый из которых содержит по меньшей мере одну из двух разных композиций. Например, такие таблетки можно изготовлять в прессе для таблеток, заполняя камеру пресса первой композицией и прессуя указанную первую композицию с последующим нанесением второй композиции поверх спрессованной первой композиции с последующим прессованием двух композиций с образованием конечной слоистой таблетки. Композицией с немедленным высвобождением может быть любая композиция, известная в данной области. Изобретение также включает использование высокомолекулярного полиэтиленоксида с молекулярной массой, по данным реологических измерений, по меньшей мере примерно 1000000 в качестве образующего матрицу вещества в производстве твердой лекарственной формы для перорального применения с пролонгированным высвобождением, содержащей активный ингредиент, который выбирают из опиоидов, для придания твердой лекарственной форме для перорального применения с пролонгированным высвобождением устойчивости к экстракции спиртом. Полиэтиленоксид можно использовать так, как это описано здесь в рамках приведенного способа с указанными препаратами или любым другим способом, принятым в данной области. Было установлено, что препараты настоящего изобретения, содержащие высокомолекулярный полиэтиленоксид, можно сплющить до толщины, составляющей примерно 15-18% от толщины несплющенной таблетки, и сплющенная таблетка во время растворения частично или практически полностью повторяет начальную форму несплющенной таблетки, несмотря на то, что эффект набухания во время растворения приводит к утолщению таблетки и уменьшению ее диаметра. Не связывая себя с теорией,можно полагать, что высокомолекулярный полиэтиленоксид обладает памятью формы и способен восстанавливать начальную форму после деформации, например, после сплющивания, в среде, которая способствует восстановлению, например, в водной среде, используемой в тестах на растворение. Принято считать, что эта способность вносит свой вклад в устойчивость к разрушению, в частности в устойчивость лекарственных форм по данному изобретению к спирту. Изобретение также включает способ лечения, при котором лекарственную форму вводят для лечения пациента при недомогании или некоторых заболеваниях, связанных с болью, и применяют лекарственную форму согласно данному изобретению для производства медикамента для лечения недомогания или конкретного заболевания у пациента, при котором необходимо избавление от конкретной боли. В одном аспекте настоящего изобретения предложена твердая лекарственная форма для перорального применения дважды в день с пролонгированным высвобождением, которая дает среднее tmax примерно через 2-6 ч, или примерно через 2,5-5,5 ч, или примерно через 2,5-5 ч после введения человеку в постоянном режиме или в качестве единичной дозы. Лекарственная форма может содержать оксикодон или его соль или гидроморфон или его соль. В одном аспекте настоящее изобретение предлагает твердую лекарственную форму для перорального применения один раз в день с пролонгированным высвобождением, которая дает среднее tmax примерно через 3-10 ч, или примерно через 4-9 ч, или примерно через 5-8 ч после введения людям в постоянном режиме или в качестве единичной дозы. Лекарственная форма может содержать оксикодон или его соль или гидроморфон или его соль. В еще одном аспекте настоящее изобретение предлагает твердую лекарственную форму с пролонгированным высвобождением для перорального применения дважды в день, которая содержит оксико- 24018311 дон или его соль в количестве примерно 10-160 мг, причем лекарственная форма после введения людям в стационарном состоянии или в виде единичной дозы дает среднюю максимальную концентрацию в плазме (Cmax) оксикодона примерно до 240 или примерно 6-240 нг/мл. В еще одном аспекте настоящее изобретение предлагает твердую лекарственную форму с пролонгированным высвобождением для перорального применения, которая содержит оксикодон или его соль в количестве примерно 10-40 мг, причем лекарственная форма после введения людям в постоянном режиме или в виде единичной дозы дает среднюю максимальную концентрацию в плазме (Cmax) оксикодона примерно 6-60 нг/мл. В следующем аспекте изобретение предлагает твердую лекарственную форму для перорального применения с пролонгированным высвобождением, которая биоэквивалентна коммерческому продуктуOxyContin. В следующем аспекте изобретение предлагает твердую лекарственную форму для перорального применения с пролонгированным высвобождением, которая биоэквивалентна коммерческому продуктуPalladone, который продается в США с 2005 г. В следующем аспекте изобретение предлагает твердую лекарственную форму для перорального применения с пролонгированным высвобождением, в которой активным ингредиентом является оксикодон гидрохлорид, причем лекарственная форма, содержащая 10 мг оксикодон гидрохлорида, при тестировании в сравнительном клиническом исследовании биоэквивалентна эталонной таблетке, содержащей 10 мг оксикодон гидрохлорида в матричной форме препарата, содержащего:h) стеарат магния: 1,25 мг/таблетку,и причем эталонную таблетку изготавливают путем следующих стадий: 1. Eudragit RS 30D и триацетин объединяют, пропуская через сито 60 меш, и тщательно перемешивают в течение примерно 5 мин или до достижения равномерного диспергирования. 2. Оксикодон HCl, лактозу и повидон помещают в чашку гранулятора с кипящим слоем/сушителя(FBD) и суспензию распыляют на порошок в кипящем слое. 3. Если нужно уменьшить размер частиц, то после распыления гранулят пропускают через сито 12. 4. Сухой гранулят помещают в миксер. 5. Одновременно расплавляют требуемое количество стеарилового спирта при температуре примерно 70 С. 6. Расплавленный стеариловый спирт добавляют к грануляту при перемешивании. 7. Воскообразный гранулят переносят в гранулятор с кипящим слоем/сушитель или дражировочный котел и дают остыть до комнатной температуры или ниже. 8. Охлажденный гранулят просеивают через сито 12. 9. Воскообразный гранулят помещают в смеситель/блендер и покрывают нужными количествами талька и стеарата магния в течение примерно 3 мин. 10. Гранулят прессуют в 125 мг таблетки на подходящем аппарате для таблетирования. Фармакокинетические параметры, такие как Cmax и tmax, AUCt, AUCinf и т.д., описывающие кривую плазмы крови, можно получить в клинических испытаниях сначала путем введения единичной дозы активного ингредиента, например оксикодона, многим тестируемым, таким как здоровые люди. Показатели плазмы крови у отдельных тестируемых затем усредняют, например, до средних значений AUC, Cmax иtmax. В контексте настоящего изобретения фармакокинетические параметры, такие как AUC, Cmax и tmax,относятся к средним значениям. Далее в контексте настоящего изобретения параметры in vivo, такие какAUC, Cmax, tmax или анальгезирующая эффективность, относят к параметрам или показателям, полученным после введения людям в постоянном режиме или в виде единичной дозы. Значение Cmax означает максимальную концентрацию активного ингредиента в плазме крови. Значение tmax показывает момент времени, при котором достигается значение Cmax. Другими словами,tmax - это момент максимальной наблюдаемой концентрации в плазме. Величина AUC (площадь под кривой) соответствует площади под кривой концентрации. ЗначениеAUC пропорционально количеству активного ингредиента, поглощенного при циркуляции крови в целом, и, следовательно, является мерой биодоступности. Значение AUCt соответствует площади под кривой "концентрация в плазме-время" от момента введения до последних измеренных концентраций в плазме и рассчитывается по правилу трапеции восходящая прямая/логарифм нисходящей линии. Значение AUCinf представляет собой площадь под кривой "концентрация в плазме-время", экстраполированной к началу координат, и его рассчитывают по формуле где C1 - последняя измеримая концентрация в плазме;z - наблюдаемая константа скорости в последней фазе;z - наклон линейной регрессии логарифмической концентрации в зависимости от временного профиля во время последней фазы;t1/2z - наблюдаемый полупериод последней фазы плазмы, который обычно определяют какt1/2z=(ln2)/z. Время запаздывания tlag определяют как момент времени непосредственно перед первой измеряемой концентрацией в плазме. Термин "здоровые люди" относится к мужчинам или женщинам среднего роста, веса и физиологических параметров, таких как кровяное давление и т.п. Здоровых людей для целей настоящего изобретения выбирают согласно критериям включения и исключения, которые основаны и соответствуют рекомендациям Международной конференции по гармонизации клинических испытаний (ICH). Таким образом, критерии включения рассматривают возраст мужчин и женщин между 18 и 50 годами включительно, массу тела в интервале 50-100 кг (110-220 фунт) и индекс массы тела (BMI) 18 и 34 (кг/м 2); такие субъекты являются здоровыми и не имеют заметных признаков отклонений, как это следует из медицинской истории, физического осмотра, жизненных показателей и электрокардиаграммы; женщины, способные к деторождению, должны использовать адекватные и надежные способы контрацепции, такие как спермицидная пена или гель, внутриматочное устройство, гормональная контрацепция(одни только гормональные контрацептивы не приемлемы); женщины, которые находятся в менопаузе,должны иметь срок менопаузы 1 года и повышенное содержание сывороточного гормона, стимулирующего фолликулы (FSH), и субъекты должны быть согласны употреблять всю пищу, которую предлагают во время испытаний. Другие критерии для включения касаются ограничений в физических упражнениях во время всего исследования и согласия не начинать новой программы тренировки и избегать необычных физических усилий. Критерии исключения включают беременных женщин (положительный тест с бета-человеческим хорионическим гонадотропином) или кормящих женщин, любой истории или современного состояния злоупотребления лекарствами или алкоголем в течение пяти лет, прошлых или любых текущих заболеваний, которые могут наложиться на прием лекарства, его распределение, метаболизм или выделение,использование опиоид-содержащих медикаментов в последние 30 суток, история известной чувствительности к оксикодону, налтрексону или родственным соединениям, любая история частой тошноты или рвоты любой этиологии, любая история постоянных приступов головной боли или травмы головы,участие в клинических испытаниях лекарств за 30 дней до первой дозы в данном испытании, любое заметное недомогание в течение 30 дней до принятия первой дозы в данном испытании, применение любых медикаментов, включая заместительную терапию тироидными гормонами (гормональная контрацепция разрешена), витамины, травы и/или минеральные добавки в течение предшествующих 7 дней перед первой дозой, отказ от воздержания от пищи в течение предшествующих 10 ч и последующих 4 ч после введения или в течение 4 ч после введения изучаемых лекарств и полный отказ от кофеина или ксантина в течение всего срока, употребления алкогольных напитков в течение 48 ч после введения первой испытуемой пробы (день 1) или в любое время, следующее за первым введением лекарства, история курения или использования никотин-содержащих продуктов в течение 45 суток до введения изучаемого лекарства или положительный тест на удержание мочи, кровь или продукты крови, употребленные в течение 30 дней до введения изучаемых лекарств или в любое время во время исследования, за исключением тех, которые нужны по клиническому протоколу, положительные результаты на лекарство в моче,пробы на спирт в любом периоде и поверхностные антигены гепатита В (HBsAg), поверхностные антитела гепатита В (HBsAb пока не иммунизированы), антитела гепатита С (анти-HCV), положительный тест на Naloxone HC1, наличие синдрома Гильберта или любых известных гепатобилиарных отклонений и заключение исследователя о том, что данный субъект не пригоден по причинам, которые конкретно не указаны выше. Субъекты, удовлетворяющие всем критериям включения и не отвечающие ни одному критерию исключения, будут приняты для исследования. Включенные субъекты представляют собой группу людей, которые дали согласие на свое участие на основе полученной информации. Рандомизированный набор субъектов представляет собой группу людей, которые отобраны случайным образом, участвуют в испытании изучаемого лекарства и в конце проходят по меньшей мере одну оценку безопасности дозы. Группа для полного анализа по показателям фармокинетического исследования (ФК) будет представлять собой группу субъектов, которые отобраны случайным образом, получают исследуемое лекарство и имеют по меньшей мере один значимый показатель ФК. Субъекты, у которых в течение 12 ч после введения дозы наступала рвота, могут быть включены в базу данных с учетом визуального обследования ФК-профиля. Субъекты и профили/метрики, исключенные из серии анализов, будут задокументированы в плане статистического анализа. Для теста на налоксон HCl сначала определяют жизненные показатели и оксиметрию пульса (SPO2). Налоксон HCl можно вводить внутривенно или подкожно. При внутривенном вливании игла или катетер должна оставаться в руке. 0,2 мг налоксона HCl (0,5 мл) вводят внутривенной инъекцией. Субъекта наблюдают в течение 30 с на признаки или симптомы для отмены. Затем вводят 0,6 мг налоксона HCl(1,5 мл) внутривенной инъекцией. Субъекта наблюдают в течение 20 мин на признаки или симптомы для отмены. При подкожном введении вводят 0,8 мг налоксона HCl (2,0 мл) и субъекта наблюдают в течение 20 мин на признаки или симптомы для отмены. Через 20 мин после инъекции налоксона проводят тест на жизненные показатели и SPO2. Жизненные показатели включают систолическое давление крови, диастолическое давление крови,частоту пульса и дыхания и температуру во рту. При опросе субъектов не задают вопрос: "как Вы себя чувствуете" при каждом обследовании, а спрашивают: "замечаете ли Вы какие-либо изменения в здоровье после исследования/после того, как Вас спрашивали последний раз" Ответ субъекта оценивают для выявления отрицательного эффекта. Субъектам также предлагают самим сообщать об отрицательных признаках в любое время во время исследования. Питание каждого субъекта будет включать стандартную пищу с высоким содержанием жира согласно "Руководству для промышленности: изучение биодоступности и биоэквивалентности пищи"Research, December 2002). Пищу дают за 30 мин до принятия дозы и ее съедают с обычной скоростью в течение 25 мин, так что заканчивают еду за 5 мин до приема дозы. Клинические лабораторные испытания, проводимые в ходе клинических исследований, включают биохимию (натощак по меньшей мере через 10 ч после еды), гематологию, серологию, анализ мочи, тест на злоупотребление лекарствами и другие тесты. Биохимические анализы (голод в течение по меньшей мере 10 ч) включают определение альбумина,щелочной фосфатазы, аланин аминотрансферазы (аланин трансаминазы, ALT), аспартат аминотрансферазы (аспартат трансаминазы, AST), кальция, хлоридов, креатинина, глюкозы, неорганического фосфата,калия, натрия, общего билирубина, общего белка, мочевины, лактат дегидрогеназы (LDH), прямого билирубина и CO2. Гематологический анализ включает определение гематокрита, гемоглобина, количества плоских клеток, количества эритроцитов, лейкоцитов, дифференциальный подсчет белых кровяных телец (% и абсолютный): базофилы, эозинофилы, лимфоциты, моноциты и нейтрофилы. Серологические исследования включают определение поверхностных антигенов гепатита В(HBsAg), поверхностных антител гепатита В (HBsAb) и антител гепатита С (анти-HCV). Анализ мочи включает определение цвета, прозрачности, рН, сахара, кетонов, уробилиногена, нитрита, содержание крови, белка, лейкоцитэстеразы, микроскопическое и макроскопическое исследование,определение удельного веса. Анализ на злоупотребление лекарствами включает анализ на опиаты, амфетамины, каннабиоиды,бензодиазепины, кокаин, котинин, барбитураты, фенциклидин, метадон и пропоксифен и спирт, например тест на спирт в крови и следы алкоголя при дыхании. Дальнейшие тесты для женщин включают только сывороточный тест на беременность, анализ мочи на беременность и тест на фолликулостимулирующий сывороточный гормон (FSH) (только для женщин в менопаузе, по их словам). Подробное описание предпочтительных вариантов Далее авторы проиллюстрируют изобретение более полно со ссылкой на сопровождающие примеры. Следует, однако, понимать, что последующее описание является только иллюстративным и никоим образом не ограничивает изобретение. Пример 1. В примере 1 таблетку 200 мг, включающую 10 мг оксикодон HCl, приготовили из высокомолекулярного полиэтиленоксида в комбинации с гидроксипропилцеллюлозой. Композиция. Способ получения. Стадии обработки для приготовления таблеток включали следующее. 1. Оксикодон HCl, полиэтиленоксид и гидроксипропилцеллюлозу смешали в сухом виде в двухлопастном низко/высокоскоростном миксере BlackDecker Handy Chopper емкостью 1,5 чашки. 2. Смесь со стадии 1 прессовали до заданной массы на однопозиционном прессе Manesty Type F 3. 3. Таблетки со стадии 2 распределяли на лотке, помещенном в сушильный шкаф Hotpack model 435304 при 70 С примерно на 14,5 ч для отверждения таблеток. Тестирование in vitro, включая тестирование на устойчивость к разрушению (тест с молотком и разрушающим усилием) и устойчивость к экстракции спиртом, проводили следующим образом. Таблетки тестировали in vitro на Аппарате USP 2 (с лопастью) при 50 об/мин в 900 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, с использованием спектрофотометраPerkin Elmer UV/VIS Lambda 20, УФ-анализ при 230 нм. Результаты представлены в табл. 1.1. Тестировали неотвержденные таблетки, отвержденные таблетки и разрушенные, т.е. сплющенные таблетки. Отвержденные таблетки сплющивали 7 ударами молотка вручную до физического разрушения. На отдельных образцах определили размеры таблеток до и после сплющивания и профили растворения. Результаты представлены в табл. 1.1. В качестве еще одного теста на устойчивость к разрушению отвержденные таблетки подвергали тесту на разрушающее усилие с приложением усилия максимум 196 Н на аппарате Schleuniger 2E/106. Результаты также представлены в табл. 1.1. Кроме того, отвержденные таблетки тестировали in vitro в среде этанол/SGF с концентрациями этанола 0, 20 и 40% для оценки экстракции этанолом. Тестирование проводили на Аппарате USP 2 (с лопастью) при 50 об/мин в 500 мл среды при 37 С, с использованием спектрофотометра Perkin Elmer UV/VISLambda 20, УФ-анализ при 220 нм. Промежутки времени для образцов составляют 0,5 и 1 ч. Результаты также представлены в табл. 1.2.n = среднее из 5 измерений; 3 196+ означает, что после воздействия максимального усилия 196 Н таблетки не разрушились; Пример 2. В примере 2 приготовили три разные таблетки по 100 мг, содержащие 10 и 20 мг оксикодон HCl, с использованием высокомолекулярного полиэтиленоксида и, необязательно, гидроксипропилцеллюлозы. Композиции. Способ приготовления. Стадии обработки в производстве таблеток включали следующее. 1. Оксикодон HCl, полиэтиленоксид и гидроксипропилцеллюлозу смешали в сухом виде в низко/высокоскоростном двухлопастном миксере BlackDecker Handy Chopper емкостью 1,5 чашки. 2. Смесь со стадии 1 прессовали до заданной массы на однопозиционном прессе Manesty Type F 3. 3. Таблетки со стадии 2 распределяли на лотке, помещенном в сушильный шкаф Hotpack model 435304 при 70-75 С примерно на 6-9 ч для отверждения таблеток. Тестирование in vitro, включая тестирование на устойчивость к разрушению (лабораторный пресс и тест на разрушающее усилие), проводили следующим образом. Отвержденные таблетки тестировали invitro на Аппарате USP 2 (с лопастью) при 50 об/мин в 500 мл модельного желудочного сока, не содержащего ферментов (SGF), при 37 С, с использованием спектрофотометра Perkin Elmer UV/VIS Lambda 20,УФ при 220 нм. Тестировали отвержденные таблетки и отвержденные сплющенные таблетки. Таблетки
МПК / Метки
МПК: A61K 9/32, A61K 31/485, A61K 9/22
Метки: дозированные, формы, фармацевтические, содержащие, опиоидный, анальгетик, оральные, устойчивые
Код ссылки
<a href="https://eas.patents.su/30-18311-ustojjchivye-oralnye-farmacevticheskie-dozirovannye-formy-soderzhashhie-opioidnyjj-analgetik.html" rel="bookmark" title="База патентов Евразийского Союза">Устойчивые оральные фармацевтические дозированные формы, содержащие опиоидный анальгетик</a>
Дозированные формы для перорального применения, содержащие прогестерон, и способы их изготовления и использования

Номер патента: 15155
Опубликовано: 30.06.2011
Авторы: Горуканти Судхир Р., Ваттиконда Чандра, Дилиберти Чарльз Е., Ахмед Салах У., Гупта Санджив К.
МПК: A61K 31/57, A61K 9/16, A61K 9/48...
Метки: дозированные, прогестерон, использования, изготовления, перорального, содержащие, формы, способы, применения
Формула / Реферат:
1. Фармацевтическая дозированная форма для перорального применения в форме порошка, содержащегося в фармацевтически приемлемой капсуле, содержащая:(a) микронизированный прогестерон в количестве от приблизительно 40 до приблизительно 70 мас.% содержимого капсулы;(b) пищевое масло в количестве от приблизительно 5 до приблизительно 25 мас.% содержимого капсулы;(c) дезинтегрант в количестве от приблизительно 5 до приблизительно 25 мас.% содержимого...
Хронотерапевтические дозированные формы

Номер патента: 8224
Опубликовано: 27.04.2007
Авторы: Хиггинз Реймонд, Баикхвал Ананд Р., Кобб Жаклин, Вудкок Пол
МПК: A61K 9/20, A61K 9/24, A61K 9/22...
Метки: дозированные, формы, хронотерапевтические
Формула / Реферат:
1. Пероральная твердая дозированная форма отсроченного высвобождения, содержащая ядро, содержащее терапевтически эффективное количество лекарственного средства, и материал отсроченного высвобождения, нанесенный прессованием на указанное ядро, причем указанный материал отсроченного высвобождения содержит гетерополисахаридную смолу и гомополисахаридную смолу, причем указанное нанесенное прессованием покрытие задерживает высвобождение указанного...
Фармацевтические лекарственные формы, содержащие липидную фазу

Номер патента: 12882
Опубликовано: 30.12.2009
Авторы: Тингвалль Пер, Хёрслеф Бенгт, Ханссон Хенри
МПК: A61K 47/14, A61K 9/02, A61K 47/44...
Метки: фармацевтические, липидную, лекарственные, содержащие, формы, фазу
Формула / Реферат:
1. Таблетка для перорального введения, содержащая липидную фазу, включающую в себя до 80 мас.% или более смеси (а) триглицерида, (б) моно- и/или диглицерида, (в) липида клеточной мембраны, (г) одного или более фармакологически активных агентов, растворенных и/или диспергированных в липидной фазе, (д) воды и/или этанола и (е) фармацевтического эксципиента в виде частиц в количестве, контролирующем поглощение. 2. Таблетка по п.1, содержащая от 1...
Дозированные формы азитромицина с пониженными побочными эффектами

Номер патента: 10110
Опубликовано: 30.06.2008
Авторы: Крю Маршалл Дэвид, Хэйджен Тимоти Артур, Хербиг Скотт Макс, Томбре Авинаш Говинд, Маккрэй Скотт Болдуин, Уэст Джеймс Блэр, Аппел Лиа Элизабет, Лайон Дэвид Кейт, Фризен Двэйн Томас, Ло Джулиан Белкнэп
МПК: A61K 9/16, A61P 31/00
Метки: дозированные, эффектами, формы, побочными, пониженными, азитромицина
Формула / Реферат:
1. Пероральная дозированная форма азитромицина, включающая: (a) эффективное количество подщелачивающего агента и (b) мультичастицы, где указанные мультичастицы включают: (i) от приблизительно 20 до приблизительно 75% азитромицина, (ii) от приблизительно 25 до приблизительно 80% глицерида, включающего глицерилмонобегенат, глицерилдибегенат и глицерилтрибегенат или их смесь, и (iii) полоксамер. 2. Пероральная дозированная форма по п.1, в которой...
Новая форма ирбесартана, способы получения указанной формы и содержащие ее фармацевтические композиции

Номер патента: 3410
Опубликовано: 24.04.2003
Авторы: Офф Кристиан, Линдрад Марк Д., Моннье Оливье, Франк Брюно, Кайэнг Сэн, Уэй Ченкоу
МПК: A61P 9/04, A61K 31/4188, C07D 403/10...
Метки: композиции, содержащие, форма, способы, указанной, получения, новая, формы, ирбесартана, фармацевтические
Формула / Реферат:
1. Кристаллическая форма ирбесартана формулы имеющая кристаллический габитус, такой что соотношение между длиной и шириной кристаллов составляет от 1:1 до 10:1, и зарядовая способность которого, измеренная с помощью трибогенерации, находится в пределах от 0 до -10 нКл/г. 2. Кристаллическая форма ирбесартана формы A, отличающаяся тем, что соотношение между длиной и шириной кристаллов составляет от 1:1 до 5:1, и зарядовая способность которой,...
Предыдущий патент: Устройство для защиты колеса транспортного средства
Следующий патент: Способ очистки и углубления русел рек от донных отложений и устройство для его осуществления
Случайный патент: Способ и реактор для получения метанола