Дефицитный по репликации модифицированный вирус осповакцины, содержащий ранний/поздний промоторный элемент, и его примененние



























Формула / Реферат
1. Дефицитный по репликации модифицированный вирус осповакцины Ankara (MVA), кодирующий по меньшей мере один антиген и/или один антигенный эпитоп, где экспрессия указанного антигена и/или эпитопа антигена находится под контролем транскрипционного элемента, содержащего по меньшей мере две копии раннего промоторного элемента, запускающего раннюю экспрессию указанного антигена и/или антигенного эпитопа, и где указанный MVA обладает способностью к репродуктивной репликации in vitro в клетках фибробластов куриных эмбрионов (CEF), но не обладает способностью к репродуктивной репликации в клеточной линии кератиноцитов человека HaCaT, в клеточной линии эмбриональной почки человека 293, в клеточной линии остеосаркомы костей человека 143В и в клеточной линии цервикальной аденокарциномы человека HeLa.
2. Дефицитный по репликации вирус по п.1, где указанный MVA представляет собой MVA-BN, депонированный в ЕСАСС под номером V00083008.
3. Дефицитный по репликации вирус по п.1 или 2, содержащий предпочтительно по меньшей мере пять копий раннего промоторного элемента.
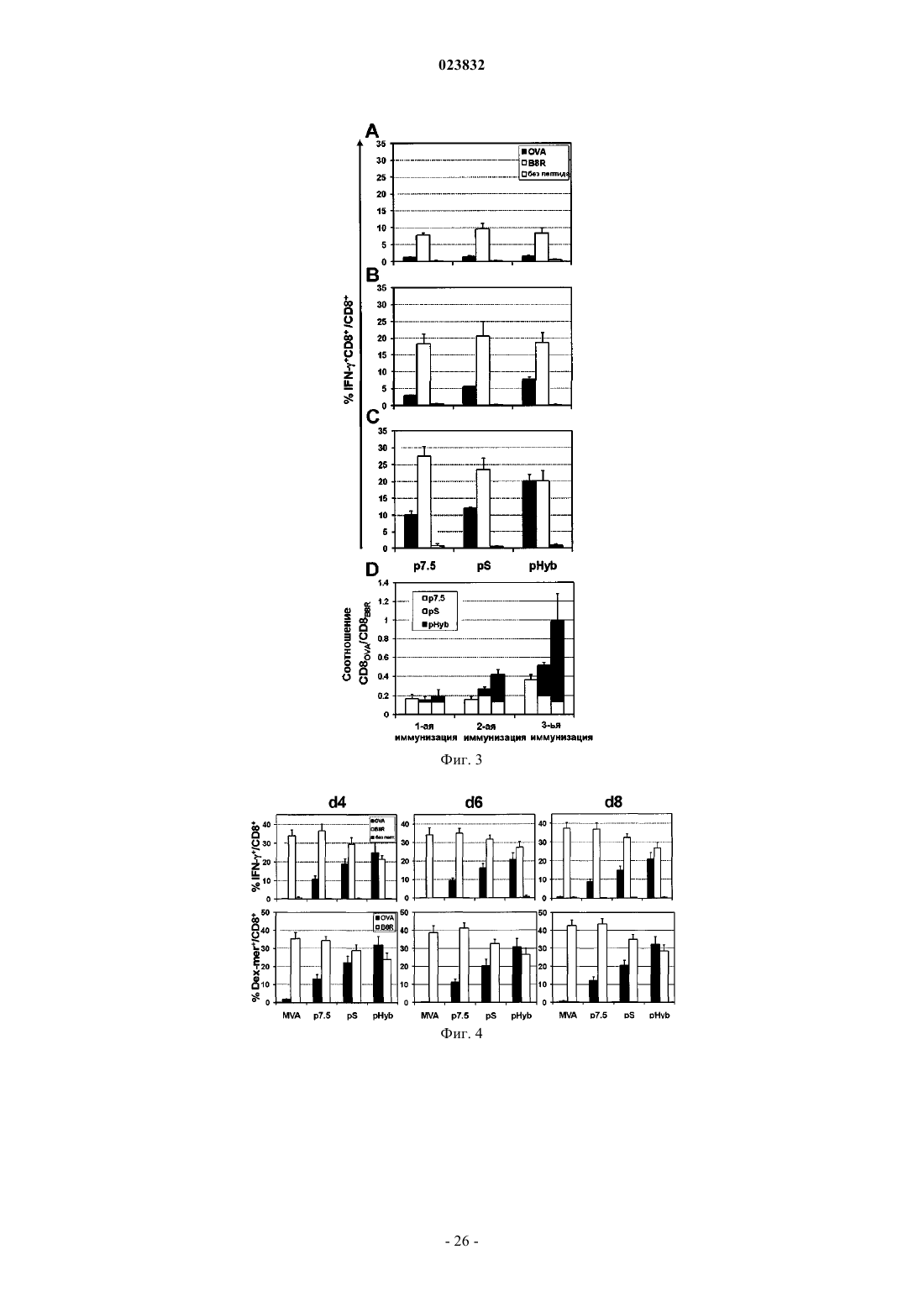
4. Дефицитный по репликации вирус по любому из предшествующих пунктов, где указанный транскрипционный элемент представляет собой или содержит ранний/поздний промоторный элемент, содержащий поздний элемент, полученный из промотора, отличного от промотора, из которого был получен ранний элемент.
5. Дефицитный по репликации вирус по п.4, где ранний/поздний промоторный элемент представляет собой гибридный ранний/поздний промоторный элемент.
6. Дефицитный по репликации вирус по п.4, где указанный поздний промоторный элемент представляет собой или содержит поздний промоторный элемент ATI вируса коровьей оспы.
7. Дефицитный по репликации вирус по любому из пп.3-6, где указанный ранний промоторный элемент представляет собой или включает ранний промоторный элемент р7.5.
8. Дефицитный по репликации вирус по п.6 или 7, где указанный ранний/поздний промоторный элемент содержит нуклеотидную последовательность SEQ ID NO:1.
9. Применение дефицитного по репликации вируса по любому из пп.1-8 в качестве лекарственного средства или вакцины для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп.
10. Применение дефицитного по репликации вируса по любому из пп.1-8 для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп.
11. Применение по п.10, где указанный T-клеточный ответ представляет собой ответ CD8 T-клеток.
12. Применение дефицитного по репликации вируса по любому из пп.1-8 для получения лекарственного средства или вакцины для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп.
13. Фармацевтическая композиция, содержащая дефицитный по репликации рекомбинантный вирус по любому из пп.1-8 и фармацевтически приемлемый носитель, разбавитель и/или адъювант.
14. Фармацевтическая композиция по п.13 для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп.
15. Фармацевтическая композиция по п.14, где указанный T-клеточный ответ представляет собой ответ T-клеток CD8.
16. Вакцина, содержащая дефицитный по репликации вирус по любому из пп.1-8 и фармацевтически приемлемый носитель, разбавитель и/или адъювант.
17. Вакцина по п.16 для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп.
18. Вакцина по п.17, где указанный T-клеточный ответ представляет собой ответ CD8 T-клеток.
19. Применение дефицитного по репликации вируса по любому из пп.1-8 при получении лекарственного средства для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп.
20. Применение по п.19, где указанный T-клеточный ответ представляет собой ответ CD8 T-клеток.
21. Применение фармацевтической композиции по п.13 для получения лекарственного средства для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп.
22. Способ индукции T-клеточного ответа в организме хозяина, в том числе человека, отличающийся тем, что указанный способ предусматривает по меньшей мере три или по меньшей мере четыре введения дефицитного по репликации вируса по любому из пп.1-8, фармацевтической композиции по любому из пп.13-15 и/или вакцины по любому из пп.16-18 указанному хозяину.
23. Способ по п.22, где ответ представляет собой CD8 T-клеточный ответ.
24. Набор, включающий по меньшей мере два флакона/контейнера для первичной/бустерной иммунизации, содержащие дефицитный по репликации вирус по любому из пп.1-8, причем первый флакон/контейнер содержит вирус для первичной иммунизации, а другой флакон/контейнер (другие флаконы/контейнеры) содержит(ат) вирус для бустерной иммунизации.
25. Ранний промоторный элемент вируса по пп.1-8, содержащий по меньшей мере два фрагмента нуклеотидной последовательности, соответствующие участку от 48 до 81 нуклеотида SEQ ID NO:1, или по меньшей мере два фрагмента нуклеотидной последовательности, которые по меньшей мере на 80% идентичны участку от 48 до 81 нуклеотида в SEQ ID NO:1.
26. Ранний промоторный элемент по п.25, дополнительно содержащий по меньшей мере один поздний промоторный элемент.
27. Ранний промоторный элемент по п.26, где промотор представляет собой поздний промоторный элемент ATI.
Текст
ДЕФИЦИТНЫЙ ПО РЕПЛИКАЦИИ МОДИФИЦИРОВАННЫЙ ВИРУС ОСПОВАКЦИНЫ, СОДЕРЖАЩИЙ РАННИЙ/ПОЗДНИЙ ПРОМОТОРНЫЙ ЭЛЕМЕНТ,И ЕГО ПРИМЕНЕННИЕ Изобретение относится к дефицитному по репликации рекомбинантному вирусу, кодирующему по меньшей мере один антиген и/или один эпитоп антигена, где экспрессия указанного антигена и/или эпитопа антигена контролируется транскрипционным элементом, содержащим по меньшей мере два элемента, запускающих раннюю экспрессию указанного антигена и/или эпитопа антигена,и к применению указанного дефицитного по репликации рекомбинантного вируса в качестве лекарственного средства или вакцины. Настоящее изобретение относится к рекомбинантному вирусу, дефицитному по репликации, который кодирует по меньшей мере один антиген и/или один эпитоп антигена, где экспрессия указанного антигена и/или эпитопа антигена контролируется транскрипционным элементом, содержащим по меньшей мере два элемента, запускающих экспрессию ранних генов указанного антигена и/или эпитопа антигена, и к применению указанного рекомбинантного вируса, дефицитного по репликации, в качестве лекарственного средства или вакцины. Предпосылки создания изобретения Вакцины, основанные на живых аттенуированных вирусах, способных к репликации, а не инактивированные препараты оказывают гораздо более эффективную защиту против вирусных инфекций и заболеваний. Указанные вакцины создают длительный и сильный иммунитет. Напротив, иммунитет, вызванный инактивированными или субъединичными вакцинами, как правило, является кратковременным. Главной причиной разработки последних является их безопасность. Обзор реплицирующихся и нереплицирующихся вирусных векторов для создания вакцин приведен в публикации Marjorie Robert-Guroff,Replicating and Non-replicating Viral Vectors for Vaccine Development, Curr. Opin. Biotechnol. 18:546-556,2007. Рекомбинантные вирусы широко используются для экспрессии чужеродных антигенов в инфицированных клетках. Так, в настоящее время проходят испытания рекомбинантных поксвирусов, которые, как полагают, являются перспективными вакцинами для индукции иммунного ответа против чужеродного антигена, экспрессированного поксвирусным вектором. Наиболее распространенными являются, с одной стороны, авипоксвирусы и вирусы осповакцины, с другой стороны - (VACV). В патентах СШАUS 5736368 и US 6051410 описан рекомбинантный вирус осповакцины штамма Wyeth, который экспрессирует антигены и белки ВИЧ. В патенте США US 5747324 описан рекомбинантный VACV штаммаNYCBH, экспрессирующий гены лентивируса. В европейском патенте ЕР 0243029 описан рекомбинантный VACV штамма Western Reserve, экспрессирующий гены ретровируса человека. Специалистам в данной области известно, что для экспрессии гетерологичных генов в поксвирусах необходимо несколько промоторов, таких как промоторы 30K и 40K (см., например, US 5747324), сильный синтетический ранний/поздний промотор (см., например, Sutter et al., A recombinant vector derived from the hosthaemagglutinin or nucleoprotein of influenza virus, J. Gen. Virol. 72,699-703, 1991) и промотор гена вируса коровьей оспы А-типа (ATI) (Li et al., High-level expression of Amsacta moorei entomopoxvirus spheroidindepends on sequences within the gene, J. Gen. Virol. 79 ,613, 1998). Все эти промоторы использовались в рекомбинантных VACV для экспрессии гетерологичных генов, и было показано, что они экспрессируют указанные гены очень эффективно и поэтому дают относительно высокие количества белка, кодируемого гетерологичным геном. В целом, при осуществлении многих стратегий иммунизации крайне желательно,чтобы антиген, против которого вызывают иммунный ответ, экспрессировался в высоких количествах. Индукция эффективного гуморального и клеточного иммунного ответа против продукта чужеродного гена, экспрессированного, например, вектором VACV, осложняется тем, что продукт чужеродного гена должен конкурировать со всеми антигенами, число которых превышает 150 вектора VACV для распознавания и индукции специфических антител и T-клеток. Иммунодоминирование эпитопов T-клетокCD8 в векторах препятствует индукции сильного ответа T-клеток CD8 против продукта чужеродного гена (Smith et al., Immunodominance of poxviral-specific CTL in a human trial of recombinant-modifiedvaccinia Ankara. J. Immunol. 175:8431-8437, 2005.) Это относится к векторам на основе реплицирующихсяVACV, в частности Dryvax, а также к дефицитным по репликации векторам, таким как NYVAC, и модифицированным вирусам осповакцины Ankara, MVA. Для экспрессии рекомбинантного антигена могут использоваться VACV поксвирусспецифичные промоторы, а не обычные эукариотические промоторы. Основанием для этого служит специфическая биология поксвирусов, которые реплицируются в цитоплазме и несут собственный, независимый от клетки аппарат транскрипции, который не распознает типичные эукариотические промоторы. Цикл репликации вируса разделяется на две основные фазы: ранняя фаза, занимающая первые 2 ч после инфицирования перед репликацией ДНК, и поздняя фаза, начинающаяся с момента репликации ДНК через 2-4 ч после инфицирования. Поздняя фаза занимает остальную часть цикла вирусной репликации и длится 2-20 ч после инфицирования перед высвобождением потомства вируса из инфицированной клетки. Существует большое число типов поксвирусных промоторов, которые различаются и обозначаются в зависимости от периода времени цикла вирусной репликации, в которой они активны,например ранний и поздний промоторы (см, например, Davison and Moss, Structure of Vaccinia Virus LatePromoter in vitro and in vivo, Journal of Virology, 64:6063-6069, 1990, все указанные статьи включены в настоящее описание в качестве ссылки). Тогда как ранние промоторы могут быть активны и на поздней стадии инфицирования, активность поздних промоторов ограничена только поздней фазой. Третий класс промоторов, который называют промежуточные промоторы, активен при переходе ранней фазы в позднюю фазу и зависит от репликации вирусной ДНК. Последнее также относится к поздним промоторам, однако транскрипция под контролем промежуточных промоторов начинается раньше, чем транскрипция под контролем обычных поздних промоторов, и требует другого набора факторов транскрипции. В последние годы стало ясно, что выбор соответствующего временного промотора поксвируса для экспрессии антигенов оказывает огромное влияние на силу и качество антигенспецифичного иммунного ответа. Было показано, что T-клеточные ответы на антигены, экспрессируемые под контролем позднего промотора, слабее, чем соответствующие ответы на те же антигены, но под контролем раннего промотора (Bronte et al., Antigen expression by dendritic cells correlates with the therapeutic effectiveness of a modelresponse. Eur. J. Immunol. 16:1479-1487, 1986). Однако еще более неожиданным оказалось то, что было показано, что при повтоноой аутологичной иммунизации с использованием VACV, а также дефицитного по репликации вектора MVA на основеVACV ответы T-клеток CD8 на антигены, находящиеся под контролем только позднего промотора, могут полностью прекратиться. Такое прекращение выражается в практически полном отсутствии антигенспецифичного ответа T-клеток CD8 после повтоноой иммунизации (Kastenmuller et al., Cross-competitionof 25 CD8+ T cells shapes the immunodominance hierarchy during boost vaccination. J. Exp. Med. 204:21872198, 2007). Таким образом, экспрессия ранних антигенов векторами VACV является, по всей видимости, важнейшим фактором для проявления эффективных антигенспецифичных ответов T-клеток CD8. Было также показано, что экспрессируемый на ранней фазе антиген вектора VACV конкурирует не только с антигенами, экспрессируемыми на поздней фазе, но также с другими ранними антигенами, что приводит к доминированию антигенной специфичности в T-клеточном CD8 ответе (Kastenmuller et al.,Cross-competition of CD8+ T cells shapes the immunodominance hierarchy during boost vaccination., J. Exp.Med. 204:2187-2198, 2007). Соответственно, специфические свойства ранней части промотора поксвируса могут быть важны для индукции антигенспецифичного T-клеточного ответа. Кроме того, хорошо известно и общепринято, что высокие уровни антигена благоприятны для индукции сильных антигенспецифичных иммунных ответов (для случая поксвирусов см., например, Wyatt et al., Correlation ofVaccine 26:486-493, 2008). Промотор, состоящий из 4 ранних промоторных элементов и элемента позднего промотора генаATI, был описан ранее, и было показано, что он контролирует повышенную экспрессию раннего антигена (Funahashi et al., Increased expression in vivo and in vitro of foreign genes directed by A-type inclusion bodyVaccine 26:486-493, 2008). T-клеточные ответы, индуцированные антигеном, экспрессия которого находится под действием такого промотора в способном к рекомбинации векторе на основе вируса осповакцины, анализировали после однократной иммунизации, и было обнаружено, что они незначительно отличаются от тех ответов, которые были получены при использовании классического промотора р 7.5 в этом же окружении (Funahashi et al., 1991). В статье Jin et al. (Constructions of vaccinia virus A-type inclusion body protein, tandemly repeatedmutant 7.5 kDa protein, and hemagglutinin gene promoters support high levels of expression, Arch. Virol. 138:315-330, 1994) была описана конструкция рекомбинантного VACV, несущего промоторы, состоящие из промотора ATI VACV в сочетании с тандемными повторами (от 2 до 38 копий) мутированного промотора р 7.5, функционально связанного с геном CAT. По-видимому, 10-15 повторов мутированного промотора р 7.5 являются эффективными для повышения экспрессии раннего гена. Однако в случае всех конструкций количество белка CAT, продуцируемого в присутствии цитозинарабинозида (AraC) (т.е., когда цикл вирусной репликации остановлен на ранней фазе), было менее одной-десятой доли от его количества, продуцируемого в отсутствие AraC, и это говорит о том, что, несмотря на то что экспрессия раннего гена была повышена, большая часть экспрессированного антигена, по-видимому, продуцировалась на поздней фазе инфекции. Таким образом, существует необходимость в улучшенных вирусных векторах, которые бы обеспечили экспрессию ранних чужеродных антигенов и вызывали индукцию эффективного антигенспецифичного иммунного ответа. Краткое описание изобретения Настоящее изобретение относится к дефицитному по репликации рекомбинантному вирусу, кодирующему по меньшей мере один антиген и/или один эпитоп антигена, где экспрессия указанного антигена и/или эпитопа антигена находится под контролем транскрипционного элемента, содержащего по меньшей мере два элемента, контролирующих раннюю экспрессию указанного антигена и/или эпитопа антигена. Настоящее изобретение также относится к указанному дефицитному по репликации рекомбинантному вирусу для применения его в качестве лекарственного средства или вакцины и к применению его для получения лекарственного средства или вакцины. В другом аспекте настоящее изобретение относится к фармацевтической композиции или вакцине,содержащей дефицитный по репликации рекомбинантный вирус и, необязательно, фармацевтически приемлемый носитель, разбавитель, адъювант и/или добавку. Настоящее изобретение также относится к указанному дефицитному по репликации рекомбинантному вирусу или к указанной фармацевтической композиции или вакцине для индукции T-клеточного ответа в организме-хозяине на указанный по меньшей мере один антиген и/или один эпитоп антигена. В другом аспекте настоящее изобретение относится к использованию указанного дефицитного по репликации рекомбинантного вируса или указанной фармацевтической композиции при получении лекарственного средства для индукции T-клеточного ответа в организме-хозяине на указанный по меньшей мере один антиген и/или один эпитоп антигена. Настоящее изобретение также относится к набору, содержащему по меньшей мере два флакона для первичной/бустерной иммунизации, содержащих указанный дефицитный по репликации рекомбинантный вирус для первой инокуляции ("первичная инокуляция") в первом флаконе/контейнере и, по меньшей мере, для второй, и/или третьей, и/или последующей инокуляции ("бустерная инокуляция") во втором и/или другом флаконе/контейнере. Настоящее изобретение также относится к способу индукции T-клеточного ответа, предпочтительно ответа T-клеток CD8, в организме-хозяине, в том числе у человека, где указанный способ предусматривает по меньшей мере три или по меньшей мере четыре введения дефицитного по репликации рекомбинантного вируса в организм хозяина. Кроме того, настоящее изобретении относится к промотору, содержащему по меньшей мере 2 элемента нуклеотидной последовательности участка нуклеотидов 48-81 в SEQ ID NO:1 и/или по меньшей мере 2 элемента нуклеотидной последовательности, которые по меньшей мере на 80% идентичны последовательности участка нуклеотидов 48-81 в SEQ ID NO:1. Подробное описание изобретения Настоящее изобретение относится к дефицитному по репликации рекомбинантному вирусу, кодирующему по меньшей мере один антиген и/или один эпитоп антигена, где экспрессия указанного антигена и/или эпитопа антигена находится под контролем транскрипционного элемента, содержащего по меньшей мере два элемента, запускающих раннюю экспрессию указанного антигена и/или эпитопа антигена. Неожиданно было обнаружено, что в случае дефицитных по репликации рекомбинантных вирусов экспрессия антигена, находящаяся под контролем транскрипционного элемента, содержащего по меньшей мере два элемента, запускающих экспрессию ранних генов, происходит значительно раньше в цикле вирусной репликации и значительно более активно в любой временной точке цикла после инфицирования, чем в случае экспрессии, находящейся под контролем стандартного транскрипционного элемента. Дополнительное преимущество заключается в высокой экспрессии ранних генов антигена, продолжающейся по меньшей мере 6 ч после инфицирования. Соответственно, в предпочтительном варианте осуществления настоящего изобретения по меньшей мере два элемента запускают экспрессию немедленно ранних генов антигена и/или эпитопа антигена, т.е. в течение первых часов после инфицирования, предпочтительно в течение первых 30 мин после инфицирования. Возможность экспрессии немедленно ранних генов антигенов под контролем транскрипционных элементов по настоящему изобретению, вытесняя ранние антигены, исследовали путем введения дефицитного по репликации рекомбинантного вируса и с последующим определением T-клеточных ответов. Иммунизация неожиданно дала высокоэффективный антигенспецифичный T-клеточный ответ, в частности ответы T-клеток CD8. Еще более удивительно, что в ряде экспериментов эта стратегия позволила полностью изменить порядок доминирования антигенной специфичности и сделать субдоминантный эпитоп T-клеток CD8 доминирующим. Этот результат не был достигнут при использовании обычных контрольных элементов транскрипции даже после четырех последовательных иммунизаций. Кроме того,антигенспецифичный ответ T-клеток CD8 через три или более цикла иммунизации вирусами по настоящему изобретению становился сильнее, если был использован стандартный контрольный транскрипционный элемент. Следует отметить, что, кроме неожиданных результатов, связанных с использованием дефицитных по репликации рекомбинантных вирусов по настоящему изобретению, отмечалось максимальное сниже-3 023832 ние вероятности побочных эффектов, таких как индукция заболевания в организме-хозяине, что дает преимущества использованию дефицитных по репликации рекомбинантных вирусов по настоящему изобретению по сравнению с компетентными по репликации рекомбинантными вирусами. В контексте настоящего описания термины "антиген" или "эпитоп антигена" используются для обозначения последовательности, которая специфически распознается или специфически связывается с компонентом иммунной системы. В основном белковый антиген отличается высоким разнообразием по размеру и распознается молекулой MHC/HLA, с которой фрагмент указанного белкового антигена связывается на антигенпрезентирующей клетке. Так, обычно термин "антиген" используется применительно к (более длинной) последовательности, в частности к (более длинной) аминокислотной последовательности или белковой последовательности, тогда как фраза "эпитоп антигена" относится к (более короткой) последовательности, в частности аминокислотному участку или пептиду, соответственно, который сохраняет способность вызывать иммунный ответ. Предпочтительно указанный антиген и/или эпитоп антигена представляет собой раковый антиген или антиген и/или эпитоп антигена инфицирующего агента, предпочтительно выбранного из вирусов,грибов, патогенных одноклеточных эукариотических и прокариотических организмов и паразитических организмов. Особенно предпочтительные примеры вирусных антигенов, которые подходят для использования в настоящем изобретении, включают антигены ретровирусов (включая HIV-1 и HTLV), герпесвирусов(включая вирус краснухи), вирусов гепатита, гепаднавирусов, вируса гриппа, пикоронавирусов (включая такие вирусы, как вирус полиомиелита), коронавирусов, буньявирусов, аренавирусов, филовирусов или из других вирусов, вызывающих геморрагическую лихорадку. Примеры предпочтительных раковых антигенов включают специфичный простатический антиген(PSA), антиген против простатической кислой фосфатазы (РАР) и Her-2/neu антигены. Предпочтительные бактериальные антигены включают антигены сибирской язвы. В контексте настоящего описания, термин "рекомбинантный вирус" относится к любому вирусу,который содержит дополнительную гетерологичную нуклеиновую кислоту, которая в природном состоянии не является частью вирусного генома, как, например, промотор по настоящему изобретению. Указанный промотор может контролировать экспрессию собственного вирусного антигена или собственного вирусного эпитопа антигена и/или может контролировать экспрессию гетерологичного или рекомбинантного гена. Гетерологичный или рекомбинантный ген может представлять собой, например,ген, кодирующий вирусный, бактериальный, грибной или раковый антиген, терапевтический ген, ген,кодирующий пептид, включающий по меньшей мере один эпитоп, который вызывает иммунный ответ. Другие примеры гетерологичного гена включают антисмысловую кассету экспрессии или ген рибозима. В контексте настоящего описания термин "дефицитный по репликации вирус" обозначает вирус, у которого снижена способность или у которого отсутствует способность к репродуктивной репликации клеток-хозяев. Предпочтительно дефицитные по репликации вирусы по настоящему изобретению включают вирусы, которые совсем не реплицируются в клетках организма-хозяина, в частности в клетках человека, и которые в этой связи являются некомпетентными по репликации. Однако в рамки настоящего изобретения включены также такие вирусы, которые демонстрируют минимальную остаточную репликативную активность, контролируемую иммунной системой организма-хозяина. Кроме того, вирусы, используемые по настоящему изобретению, предпочтительно способны инфицировать клетки-хозяева, но по существу не способны или вовсе не способны продуцировать инфекционное вирусное потомство в инфицированных клетках. Вирусы, которые "способны инфицировать клетки" представляют собой вирусы, на вирусной поверхности которых расположены структуры, способные взаимодействовать с клетками-хозяевами в такой степени, чтобы вирус или, по меньшей мере, вирусный геном мог проникать в клетку-хозяина. В контексте настоящего описания термин "вирус, не способный продуцировать инфекционное потомство вируса в указанных клетках", относится к вирусам, геном которых, по меньшей мере частично,транскрибируется и транслируется в вирусные белки или даже реплицируется, однако не упаковывается с образованием инфекционных вирусных частиц. Таким образом, вирусы, используемые по настоящему изобретению, представляют собой вирусы, ведущие к абортивным инфекциям хозяина. Абортивные инфекции могут происходить по двум причинам. Согласно первой альтернативе клетка может быть чувствительна к инфекции, но может не разрешать размножение вируса, например, из-за того, что не все вирусные гены экспрессированы в форме,необходимой для размножения вируса в указанной клетке. Примером такого типа вируса по настоящему изобретению для клеток человека является модифицированный вирус осповакцины Ankara (MVA), который более подробно описан ниже. В соответствии со второй альтернативой абортивная инфекция может быть результатом инфекции клеток дефектными вирусами, которые утратили полный набор вирусных генов. Примером такого вируса по настоящему изобретению для клеток человека является DISC-HSV1 (дефектный вирус простого герпеса с одним циклом), т.е. вирус простого герпеса, который ограничен одним циклом инфицированияhematopoietic cells, Blood, 89:119-5, 127, 1997). Такой вирус не имеет гена для обязательного гликопротеина Н (gH), но может расти, давая высокие титры в комплементарной клеточной линии, экспрессирующей gH. В некомплементарных клеточных линиях, в которых рост вируса простого герпеса невозможен, указанный вирус ограничен одним циклом репликации, ведущим к высвобождению неинфекционного вируса. Вирусы по настоящему изобретению предпочтительно реплицируются по меньшей мере в одном типе клеток и по меньшей мере одного вида животных. Соответственно, вирус можно амплифицировать перед введением в клетку-хозяина для вакцинации и/или лечения. В качестве примера можно привестиMVA, который может амплифицироваться в клетках CEF, но не способен образовывать инфекционное потомство вируса в клетках человека. Предпочтительные примеры дефицитных по репликации вирусов, подходящих для использования в настоящем изобретении, включают вирусы аденовирусной, герпесвирусной и поксвирусной природы. Примеры дефицитного по репликации аденовируса, подходящего для использования в настоящем изобретении, включают Е 1-дефицитный дефектный по репликации аденовирус человека, описанный в статье Sharpe et al., Single oral immuinization with replication deficient recombinant adenovirus elicits long-livedtransgene-specific cellular and humoral immune response, Virology, 293, 210-216, 2002. Пример дефицитного по репликации вируса герпеса, подходящего для использования в настоящем изобретении, включаетDISC-HSV1, которые выше уже были указаны. В предпочтительном варианте осуществления настоящего изобретения дефицитный по репликации рекомбинантный вирус представляет собой поксвирус, такой как, например, авипоксвирус или ортопоксвирус, такие как вирусы осповакцины. Примеры авипоксвирусов, подходящих для использования в настоящем изобретении, включают любой авипоксвирус, такой как Fowlpoxvirus, Canarypoxvirus, Uncopoxvirus, Mynahpoxvirus,Pigeonpoxvirus, Psittacinepoxvirus, Quailpoxvirus, Peacockpoxvirus, Penguinpoxvirus, Sparrowpoxvirus,Starlingpoxvirus и Turkeypoxvirus. Предпочтительные авипоксвирусы включают Canarypoxvirus и Fowlpoxvirus. Авипоксвирусы по своей природе ограничены хозяином и возможностью репликативной продукции только в видах птиц и их клетках (Taylor et al., Biological and immunogenic properties of acanarypox-rabies recombinant, ALVAC-RG (vCP65) in non-avian species, Vaccine, 13:539-549, 1995). Если клетки человека инфицируются авипоксвирусом, гетерологичные гены экспрессируются из вирусного генома. Однако авипоксвирус не реплицируется в клетках человека, и, соответственно, у человека отсутствует риск повреждения репродуктивной репликацией вируса. Были сконструированы различные рекомбинантные авипоксвирусы, которые экспрессируют, например, продукты генов лентивируса(US 5766598), цитокины и/или опухолеассоциированные антигены (US 5833975) или гликопротеин G вируса бешенства (Taylor et al., Biological and immunogenic properties of a canarypox-rabies recombinant,ALVAC-RG (vCP65) in non-avian species, Vaccine, 13:539-549, 1995). С рекомбинантным канарипоксвирусом, экспрессирующим четыре гена ВИЧ: gag, pol, env и nef, уже проводятся клинические испытания(Peters, В.S., The basis for HIV immunotherapeutic vaccines, Vaccine, 20:688-705, 2001). Поскольку авипоксвирусы реплицируются только в клетках птиц, то эти клетки используют для амплификации вируса и для получения рекомбинантных вирусов. Примером канарипоксвируса является штамм Rentschler. Выделенный из бляшки и очищенный штамм Canarypox, обозначенный как ALVAC (US 5766598), был депонирован в соответствии с условиями Будапештского договора в American Type Culture Collection (ATCC), под номером VR-2547. Другой штамм Canarypox представлен в коммерчески доступной канарипоксвирусной вакцине, где имеющийся штамм канарипокса, обозначенный как LF2 СЕР 524241075, доступен от Institute Merieux, Inc. Примеры Fowlpoxvirus включают штаммы FP-1, FP-5 и TROVAC (US 5766598). FP-1 представляет собой штамм Duvette, модифицированный для использования его в качестве вакцины у однодневных цыплят. Указанный штамм, включенный в коммерчески доступную фаулпоксвирусную вакцину, был обозначен как 0 DCEP 25/СЕР 67/2309, октябрь 1980 г. и доступен от Institute Merieux, Inc. FP-5 представляет собой коммерчески доступный штамм фаулпоксвируса вакцины из эмбриона цыпленка, доступный от American Scientific Laboratories (Division of Schering Corp.) Madison, Wisconsin, ветеринарная лицензия США 165, серийный 30321 (United States Veterinary License No. 165, serial No. 30321). В особенно предпочтительном варианте осуществления настоящего изобретения дефицитный по репликации рекомбинантный вирус представляет собой ортопоксвирус, такой как вирус осповакцины. Примеры вирусов осповакцины, подходящие для использования в настоящем изобретении, включают вирус осповакцины, штамм DIs, который хорошо растет в CEF, но не способен расти в большинстве клеток млекопитающих (Tagaya et al., A new mutant of dermovaccinia virus, Nature Vol. 192, No. 4800, 381383, 1961; Ishii et al., Structural analysis of vaccinia virus DIs strain: Application as a new replication deficientviral vector, Virology 302, 433-444, 2002). Другой предпочтительный пример подходящего вируса осповакцины представляет собой высокоаттенуированный штамм вируса осповакцины NYVAC, который был получен из клонированного из бляшки изолята вакцинного штамма Copenhagen путем делеции 18 ORF(открытых рамок считывания) из вирусного генома (Tartaglia et al., NYVAC: A highly attenuated strain ofvaccinia virus, Virology, 188, 217-232, 1992). Штамм NYVAC отличается резким снижением способности к репликации в различных культурах клеток ткани человека, но сохраняет способность индуцировать мощные иммунные ответы против внешних антигенов. Хотя ниже настоящее изобретение будет описано более подробно для рекомбинантных вирусов осповакцины, таких как рекомбинантный MVA, все указанные выше вирусы также подходят для использования в настоящем изобретении. В предпочтительном варианте осуществления настоящего изобретения указанный дефицитный рекомбинантный вирус представляет собой рекомбинантный модифицированный вирус осповакциныMVA относится к вирусу осповакцины (Vaccinia virus) - представителю рода Orthopoxvirus семейства Poxviridae. MVA был получен путем более 570 серийных пассажей в фибробластах эмбрионов цыпленка из кожного штамма осповакцины Ankara (Chorioallantois vaccinia virus Ankara virus, CVA; обзор см., например, Mayr, A., et al., Passage History: Abstammung, Eigenschaften und Verwendung des attenuiertenVaccinia-Stammes MVA, Infection 3, 6-14, 1975), который поддерживается в Институте вакцинации, Анкара, Турция (Vaccination Institute, Ankara, Turkey) в течение многих лет и используется в качестве основы для вакцинации людей. Однако в связи с наличием частых тяжелых поствакцинационных осложнений, связанных с вирусами осповакцины, были предприняты неоднократные попытки получить более ослабленный и более безопасный вирус оспы человека. В период с 1960 по 1974 гг. профессор AntonMayr смог получить аттенуированнный вирус CVA в результате 570 непрерывных пассажей в клеткахVaccinia-Stammes MVA. Infection 3: 6-14, 1975). На множестве моделей животных было показано, что полученный штамм MVA является авирулентным (Mayr, A.Danner, K. Vaccination against pox diseasesMVA исследовали в рамках клинических испытаний в качестве вакцины для иммунизации против оспы человека (Mayr et al., Zbl. Bakt. Hyg. I, Abt. Org. В 167, 375-390 [1987], Stickl et al., MVA vaccinationWschr. 99, 2386-2392, 1974). На ранних стадиях разработки MVA в качестве предварительной противооспенной вакцины, проводились клинические испытания с использованием MVA-517 (который соответствует 517-ому пассажу) в сочетании с Lister Elstree (Stickl, Smallpox vaccination and its consequences: first experiences with the highlyattenuated smallpox vaccine "MVA". Prev.Med. 3(1): 97-101, 1974; Stickl and Hochstein-Mintzel, Intracutaneous smallpox vaccination with a weak pathogenic vaccinia virus ("MVA virus"). Munch Med Wochenschr. 113: 1149-1153, 1971) у индивидуумов с риском развития побочных реакций при применении вируса осповакцины. В 1976, MVA, полученный из посевного материла MVA-571 (соответствует 571-ому пассажу), был зарегистрирован в Германии в качестве первой вакцины для проведения программы двухстадийной парентеральной вакцинации человека против оспы. Позднее, MVA-572 использовали у более 120000 индивидуумов европеоидной расы, в основном это были дети в возрасте от 1 до 3 лет, и не было зарегистрировано ни одного побочного эффекта, даже несмотря на то, что многие из этих индивидуумов в исследуемой популяции имели высокий риск развития осложнений, связанных с использованием обычного вируса осповакцины (Mayr et al., 1978, The smallpox vaccination strain MVA: marker, genetic structure,experience gained with the parenteral vaccination and behaviour in organisms with a debilitated defencemechanism (author's transl). Zentralbl. Bacterid. (B) 167:375-390). MVA-572 был депонирован в Европейской Коллекции культур клеток животного происхождения (European Collection of Animal Cell Cultures) под номером ЕСАСС V94012707. MVA обладал сниженной вирулентностью, при том что сохранялась хорошая иммуногенность. Поскольку при аттенуировании MVA проводили большое число пассажей, имеется большое число штаммов или изолятов, в зависимости от количества пассажей в клетках CEF. Все штаммы MVA взяты у доктора Mayr, и большая часть из них получена из MVA-572, который использовался в Германии в рамках программы борьбы с оспой, или из MVA-575, который широко использовался в качестве вакцины в ветеринарии. MVA-575 был депонирован 7 декабря 2000 г. в Европейской Коллекции культур клеток животных (European Collection of Animal Cell Cultures (ЕСАСС с номером доступа V00120707. ПродуктMVA-BN, использованный, в качестве характерного примера, для создания рекомбинантного MVA по настоящему изобретению, был получен из MVA-584 (соответствует 25-58-му пассажу MVA в клеткахCEF). Образец MVA-BN был депонирован 30 августа 2000 г. в Европейской Коллекции культур клеток(European Collection of Cell Cultures (ЕСАСС с номером доступа V00083008. В результате длительных пассажей родительского вируса осповакцины Ankara (CVA) в хориоаллантоис геном полученного вируса MVA содержал делеции размером примерно 27 тыс.п.н. в геномной последовательности и в этой связи был описан как ограниченный, главным образом, клетками птиц, используемыми в качестве клеток-хозяев (Meyer, H. et al., Mapping of deletions in the genome of the highlyattenuated vaccinia virus MVA and their influence on virulence, J. Gen. Virol. 72, 1031-1038, 1991). Аттенуированные штаммы утратили приблизительно 13% (примерно 26,5 т.п.н. за счет делеции шести основных и множества минорных сайтов) из кодирующего участка генома по сравнению с вирусомпредшественником CVA (Meisinger-Henschel et al., Genomic sequence of chorioallantois vaccinia virusAnkara, the ancestor of modified vaccinia virus Ankara, J. Gen. Virol. 88, 3249-3259, 2007). Делеции влияют на множество генов вирулентности и целый диапазон генома хозяина, а также затрагивают крупный фрагмент гена кодирующего участка белка включения А-типа (ATI) и гена, кодирующего структурный белок, направляющий частицы зрелого вируса в тела включения А-типа. Таким образом, настоящее изобретение относится к дефицитным по репликации рекомбинантнымMVA вирусам, полученным на основе любого и всех MVA вирусов. Соответственно, штамм MVA с номером VR-1508, депонированный в Американской Коллекции типовых культур (American Type Culturecollection (ATCC), Manassas, VA 20108, USA), а также указанные выше штаммы MVA вируса, в частности штаммы MVA 572 и 575, депонированные в Европейской Коллекции культур клеток животныхECACC V00120707 соответственно, являются предпочтительными штаммами по настоящему изобретению. Особенно предпочтительные MVA вирусы включают MVA, штаммы MVA-BN, такие, например,как штаммы, депонированные в ЕСАСС с номером V00083008, и их варианты и производные, обладающие такими же свойствами, как и MVA-BN.MVA-BN может прикрепляться к клеткам человека и проникать в них, где кодируемые вирусом гены будут крайне эффективно экспрессироваться. Однако сборка и высвобождение потомства вируса не происходит. Препараты MVA-BN и его производные вводили животным 20 типов и более чем 2000 людей, в том числе индивидуумам с иммунодефицитом. Результаты показали, что все случаи вакцинации были в основном безопасными и хорошо переносились. Часть личных публикаций наводит на мысль о том, что все штаммы MVA очень близки и отражают высокоаттенуированный, безопасный, живой вектор. Однако, как показали доклинические исследования,MVA-BN обладает наилучшей степенью аттенуации и эффективностью по сравнению с другими штаммами MVA (WO 02/42480). Варианты штамма MVA, MVA-BN, такие как, например, депонированные в ЕСАСС под номером V00083008, способны к репродуктивной репликации in vitro в фибробластах куриных эмбрионов (CEF), но не демонстрируют способности к репликации в клетках человека, гдеMVA 575 или MVA 572 может репродуктивно реплицироваться. Например, MVA-BN не обладает способностью к репродуктивной репликации в клеточной линии кератиноцитов человека HaCaT, клеточной линии эмбрионов почки человека 293, клеточной линии остеосаркомы костей человека 143 В и в клеточной линии аденокарциномы шейки матки человека HeLa. Кроме того, штаммы MVA-BN не реплицируются на модели мыши, которая не способна продуцировать зрелые В- и T-клетки и которая в этой связи характеризуется крайне пониженным иммунитетом и отличается высокой чувствительностью к реплицирующемуся вирусу. Дополнительным или альтернативным качеством штаммов MVA-BN является их способность индуцировать, по меньшей мере по существу, тот же уровень иммунного ответа при использовании режимов первичной иммунизации вирусом осповакцины/бустерной иммунизации вирусом осповакцины,по сравнению с режимами,предусматривающими проведение ДНКпримирования/бустерной иммунизации вирусом осповакцины. Таким образом, в предпочтительном варианте штамм MVA по настоящему изобретению обладает способностью к репродуктивной репликации in vitro в фибробластах куриных эмбрионов (CEF), но не способен к репродуктивной репликации в клетках человека, в которых MVA 575 или MVA 572 может репродуктивно реплицироваться. Более предпочтительно штамм MVA не способен к репродуктивной репликации в клеточной линии кератиноцитов HaCaT человека, клеточной линии эмбрионов почки человека 293, клеточной линии остеосаркомы костей человека 14 3 В и в клеточной линии аденокарциномы шейки матки человека HeLa. Таким образом, в наиболее предпочтительном варианте осуществления настоящего изобретения штамм MVA, используемый в настоящем изобретении, представляет собой штаммMVA-BN, депонированный в ЕСАСС под номером V00083008, а также его производные и варианты,проявляющие такие же свойства, которые были описаны для MVA-BN соответственно. Характерные особенности MVA-BN, описание соответствующих биологических тестов, используемых для определения, является ли данный штамм MVA штаммом MVA-BN или его производным, а также методы, позволяющие получить MVA-BN или MVA со свойствами MVA-BN, описаны вWO 02/42480. В указанном документе также описывается, как можно размножать MVA и другие вирусы осповакцины. В общих чертах эта процедура состоит в том, что эукариотические клетки инфицируют вирусом. Эукариотические клетки представляют собой такие клетки, которые характеризуются чувствительностью к инфекции соответствующим поксвирусом и позволяют осуществить репликацию и продукцию инфекционного вируса. Для MVA характерные примеры клеток такого типа включают фибробласты куриного эмбриона (CEF) и клетки BHK (Drexler et al., Highly attenuated modified vaccinia хорошо известных специалистам в данной области. Предпочтительно клетки CEF культивируют в бессывороточной среде в неподвижных колбах или роллер-флаконах. Инкубация предпочтительно проводится в течение 48-96 ч при температуре 37 С. Используемый для инфицирования MVA берут с показателем множественности инфекции (MOI) от 0,05 до 1 TCID50 и инкубацию предпочтительно проводят в течение периода времени от 48 до 72 ч при температуре 37 С. Термин "не способный к репродуктивной репликации" используется в настоящем описании согласно определению, приведенному в WO 02/42480 и в патенте США 6761893, которые включены в настоящее описание в качестве ссылок. Таким образом, указанный термин может быть использован к вирусу,который обладает способностью к вирусной амплификации в течение 4 дней после инфицирования на уровне, менее чем 1, по данным тестов, приведенных в патенте США 6761893, где описание указанных тестов включено в качестве ссылки. Термин "коэффициент амплификации" применительно к вирусу представляет собой показатель, определяемый отношением количества вируса, продуцируемого из инфицированной клетки (выход вируса), к тому количеству вируса, которое изначально использовалось для инфицирования клеток (вход вируса). Показатель соотношения "1" между количествами вируса на "выходе" и "входе" определяет статус амплификации, где количество вируса, продуцируемого из инфицированных клеток, равно количеству вируса, изначально использованного для инфицирования клеток.MVA-BN или его производные характеризуются согласно одному из вариантов осуществления настоящего изобретения способностью к индукции, по меньшей мере по существу, такого же уровня иммунитета в режимах первичной иммунизации вирусом осповакцины/бустерной иммунизации вирусом осповакцины по сравнению с режимами ДНК-примирования/бустерной иммунизации вирусом осповакцины. Вирус осповакцины рассматривается как индуцирующий, по меньшей мере по существу, такой же уровень иммунитета в режимах первичной иммунизации вирусом осповакцины/бустерной иммунизации вирусом осповакцины, по сравнению с режимами ДНК-примирования/бустерной иммунизации вирусом осповакцины, если ответ CTL, определяемый согласно любому из тестов: "тест 1" и "тест 2", описанных в WO 02/42480, предпочтительно по результатам обоих тестов, равен, по меньшей мере по существу, той же величине, что и в режимах первичной иммунизации вирусом осповакцины/бустерной иммунизации вирусом осповакцины, по сравнению с режимами ДНК-примирования/бустерной иммунизации вирусом осповакцины. Более предпочтительно ответ CTL первичной иммунизации вирусом осповакцины/бустерной иммунизации вирусом осповакцины выше, по данным по меньшей мере одного из тестов,чем в режимах ДНК-примирования/бустерной иммунизации вирусом осповакцины. Наиболее предпочтительно указанный ответ CTL выше, по данным обоих тестов. В WO 02/42480 описано получение вирусов осповакцины, имеющих свойства MVA-BN. Высокоаттенуированный вирус MVA-BN может быть получен, например, путем проведения дополнительных пассажей модифицированного вируса осповакцины Ankara (MVA), такого как MVA-572 или MVA-575,и, необязательно, путем проведения дополнительных(ой) стадий (и) очистки бляшек. В общих чертах было показано, что MVA-BN обладает наивысшим профилем аттенуации, по сравнению с другими штаммами MVA, и безопасен даже для животных с крайне ослабленным иммунитетом. Хотя репликация MVA сильно ограничена в клетках млекопитающих, гены MVA эффективно транскрибируются, но при этом репликация вируса блокируется на стадии сборки и выхода (Sutter andVirology 238:198-211, 1997.) Несмотря на высокую степень аттенуации и сниженную вирулентностьMVA-BN, в доклинических исследованиях было показано, что этот вирус вызывает как гуморальный,так и клеточный ответы на VACV и на продукты гетерологичных генов, клонированных в геноме MVAa phase I trial in metastatic melanoma. Clin Cancer Res. 10(16):5381-5390, 2004). Вакцины на основе MVA-BN и рекомбинантного MVA-BN могут быть разработаны, пассированы, получены и промышленно произведены в клетках CEF, культивируемых в бессывороточной среде. Большое число вариантов на основе рекомбинантного MVA-BN были описаны для проведения доклинических и клинических разработок. При этом не было выявлено различий в аттенуации (потери способности к репликации в линиях клеток человека) или безопасности (доклинические и клинические данные о токсичности) у MVA-BN, вектора с вирусной основой и у различных вакцин на основе рекомбинант-8 023832 ных MVA. Согласно настоящему изобретению дефицитные по репликации рекомбинантные вирусы содержат антиген и/или антигенный эпитоп, где экспрессия указанного антигена и/или антигенного эпитопа соответственно регулируется контрольным элементом транскрипции. В контексте настоящего изобретения контрольные элементы или последовательности транскрипции представляют собой регуляторные последовательности ДНК, такие как промоторные последовательности для связывания с РНК-полимеразой, энхансеры, последовательности инициации транскрипции, для связывания с рибосомой, и/или терминаторы и т.п., которые обеспечивают экспрессию интересующего антигена в клетке-хозяине. В предпочтительном варианте осуществления дефицитный по репликации рекомбинантный вирус содержит контрольный транскрипционный элемент, который содержит по меньшей мере два элемента,направляющих раннюю экспрессию интересующего антигена и/или антигенного эпитопа. Указанные по меньшей два элемента могут представлять собой промоторные элементы, предпочтительно ранние промоторные элементы, более предпочтительно по меньшей мере две и наиболее предпочтительно по меньшей мере пять копий раннего промоторного элемента. В контексте настоящего описания термины "ранний промотор" или "ранний промоторный элемент" относятся к промоторам, которые активны в инфицированных вирусом клетках, до начала репликации вирусной ДНК. Способы, позволяющие определить, является ли промотор ранним промотором, хорошо известны специалистам в данной области. В частности, интересующий промотор может быть встроен выше репортерного гена, и указанная конструкция может быть введена в вирусный вектор, например, в вектор на основе осповакцины, который затем используют для инфицирования клеток. Для оценки активности промотора в качестве раннего промотора, клетки инкубируют с веществом, которое ингибирует репликацию вирусной ДНК, например с AraC. Репликация ДНК является необходимым, заранее известным условием для активности позднего промотора. Таким образом, любая промоторная активность, определяемая в такой системе тестирования, обусловлена элементами, активными в качестве раннего промотора. Следовательно, термин "поздний промотор" относится к любому промотору, который становится активным после того, как началась репликация ДНК. Поздняя активность также может быть определена способами, хорошо известными специалистам в данной области. Для простоты изложения термин "поздний промотор" в контексте настоящего описания относится к промотору, который активен только, если не добавляют вещество, блокирующее репликацию ДНК. В предпочтительном варианте осуществления дефицитный по репликации рекомбинантный вирус содержит ранний/поздний промотор, предпочтительно ранний/поздний промотор поксвируса. Ранний/поздний промотор, осуществляющий экспрессию присоединенной последовательности нуклеиновой кислоты как на ранней, так и на поздней стадиях жизненного цикла вируса. Предпочтительно указанный ранний/поздний промотор содержит по крайней мере один поздний промоторный элемент, связанный по меньшей мере с 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 или большим числом копий раннего промоторного элемента. Еще более предпочтительно указанный ранний/поздний промотор содержит поздний промоторный элемент и по меньшей мере две, предпочтительно по меньшей мере пять копий раннего промоторного элемента, предпочтительно копий элемента из нуклеотидной последовательности, которая соответствует нуклеотидам 48-81 в последовательности SEQ ID NO:1. В другом предпочтительном варианте осуществления по меньшей мере два элемента контрольного транскрипционного элемента, в частности по меньшей мере две копии, предпочтительно по меньшей мере пять копий раннего промоторного элемента оптимизированы по последовательности. В другом предпочтительном варианте осуществления указанный ранний/поздний промотор представляет собой гибридный ранний/поздний промотор, содержащий поздний элемент, полученный из промотора, отличного от промотора, из которого был получен ранний элемент. Авторы настоящего изобретения неожиданно обнаружили, что контрольный транскрипционный элемент, осуществляющий экспрессию антигена как можно рано и как можно стабильно, дает антигену временные и количественные преимущества по сравнению с большинством аутохтонных векторных антигенов, что благоприятно для индукции сильного антигенспецифичного T-клеточного ответа, в частности ответа T-клеток CD8. Сильный ранний промотор был обозначен как MVA и использовался в качестве примера и как предпочтительный вариант дефицитного по репликации рекомбинантного вектора по настоящему изобретению. Для создания сильного раннего промотора использовали сочетание множества ранних промоторных элементов в тандемном расположении для усиления экспрессии именно на ранней фазе цикла репликации вируса. Указанный промоторный элемент объединяли с коротким поздним промоторным элементом, полученным из промотора ATI коровьей оспы, который предположительно осуществляет экспрессию гена в поздней фазе и ведет к дополнительному повышению количества экспрессированного антигена. Кинетика экспрессии сдвигалась в сторону более ранней фазы при использовании промотора, принадлежащего к недавно идентифицированному классу немедленно ранних промоторов. Немедленно ранние гены определяются как экспрессируемые в периоде, начинающиеся через 0,5-1 ч после инфицирования (Assarsson et al., Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class ofearly promoters. J. Mol. Biol. 210:749-769, 1989). Контрольный транскрипционный элемент по настоящему изобретению представляет собой наиболее предпочтительно немедленно ранний контрольный транскрипционный элемент, созданный при объединении по меньшей мере двух, предпочтительно пяти или даже мультимерных контрольных элементов транскрипции, которые предпочтительно оптимизированы по последовательности и расположены тандемно. Указанный ранний контрольный транскрипционный элемент предпочтительно сконструирован с использованием раннего промоторного элемента, более предпочтительно раннего промоторного элемента поксвируса. Предпочтительно указанный ранний промоторный элемент представляет собой ранний промоторный элемент р 7.5, наиболее предпочтительно оптимизированный по последовательности ранний промоторный элемент р 7.5. Предпочтительно ранний/поздний промотор поксвируса содержит по меньшей мере один поздний промоторный элемент, связанный по меньшей мере с 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 или большим числом копий раннего промоторного элемента. Еще более предпочтительно указанный ранний/поздний промотор поксвируса содержит поздний промоторный элемент и по меньшей мере две копии, предпочтительно по меньшей мере пять копий раннего промоторного элемента, предпочтительно копии элемента нуклеотидной последовательности, которая соответствует нуклеотидам 48-81 последовательности SEQ ID NO:1. Особенно предпочтительным является ранний/поздний промотор поксвируса, содержащий по меньшей мере две копии, предпочтительно по меньшей мере пять копий раннего промоторного элемента р 7.5, более предпочтительно раннего промоторного элемента р 7.5, оптимизированного по последовательности. Предпочтительно ранний/поздний промотор поксвируса представляет собой гибридный ранний/поздний промотор поксвируса, содержащий поздний элемент, полученный из промотора, отличного от промотора, из которого был получен ранний элемент. Согласно другому предпочтительному варианту осуществления указанный поздний элемент гибридного раннего/позднего промотора поксвируса представляет собой или содержит поздний промоторATI коровьей оспы. Предпочтительно указанный гибридный ранний/поздний промотор поксвируса содержит по меньшей мере один поздний промоторный элемент, предпочтительно промоторный элемент ATI, связанный с 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 или большим числом копий раннего промоторного элемента,предпочтительно раннего промоторного элемента р 7.5 и наиболее предпочтительно раннего промоторного элемента р 7.5, оптимизированного по последовательности. Особенно предпочтительным является дефицитный по репликации рекомбинантный вирус, как определенно выше, где указанный гибридный ранний/поздний промотор поксвируса содержит нуклеотидную последовательность SEQ ID NO:1. Последовательность промотора гена белка включения А-типа вируса коровьей оспы (промотор ATI) известна специалистам в данной области. В этой связи приводится ссылка на номер доступа D00319Genebank. Предпочтительная последовательность промотора ATI приведена ниже: 5'GTTTT GAATA AAATT TTTTT АТААТ АААТ 3' (SEQ ID NO:6). Согласно настоящему изобретению можно использовать определенный выше промотор ATI или использовать производное промотора ATI, который может быть описан подпоследовательностью, как показано выше. Термин "подпоследовательность" относится к укороченным фрагментам показанной выше последовательности, которая сохраняет активность промотора, в частности, в качестве позднего промотора вируса осповакцины. Длина типичного фрагмента последовательности промотора ATI составляет меньшей мере 10 нуклеотидов, более предпочтительно по меньшей мере 15 нуклеотидов, еще более предпочтительно по меньшей мере 20 нуклеотидов и наиболее предпочтительно по меньшей мере 25 нуклеотидов последовательности промотора ATI. Подпоследовательность предпочтительно может содержать нуклеотиды 25-29 в последовательности ATI, т.е. последовательность 5'-TAAAT-3', расположенную на 3'-конце промоторной последовательности ATI. Подпоследовательность может также содержать нуклеотиды 22-2 9 в промоторной последовательности ATI, т.е. последовательность 5-TAATAAAT-3', расположенную на 3'-конце промоторной последовательности ATI. Ранний элемент промотора р 7.5 был оптимизирован путем проведения замены одного нуклеотида,как описано ниже более подробно. Оптимизация дала промотор с повышенной экспрессией в присутствии AraC, по сравнению с вариантом в котором отсутствует AraC, в клетках HeLa. Кроме того, экспрессия повышенных количеств GFP (eGFP), осуществляемая промотором, содержащим пять копий оптимизированного раннего промоторного элемента р 7.5, связанного с одной копией позднего промотора ATI(далее обозначаемого как "промотор pHyb"), происходит не только значительно раньше, но также на значительно более высоком уровне, чем экспрессия, осуществляемая хорошо известным синтетическим промотором pS и промотором р 7.5 в период времени 30-120 мин после инфицирования. Значительные количества eGFP были обнаружены уже через 30 мин после инфицирования. Это происходило в 2-3 раза быстрее, чем было обнаружено для раннего/позднего промотора pS или р 7.5. Сочетание по меньшей мере трех раундов иммунизации с таким немедленно ранним промотором, для экспрессии антигена, приводило к повышению антигенспецифичного ответа T-клетками CD8, по сравнению с обычно используемым поксвирусным промотором р 7.5 и pS. Преимущество такой усиленной ранней экспрессии антигена длилось по меньшей мере до 6 ч после инфицирования. Таким образом, ранняя экспрессия гена была чрезвычайно высокой при использовании этого промотора. В предпочтительных вариантах осуществления рекомбинантный MVA экспрессирует высокие количества кодируемого антигена и/или эпитопа антигена на немедленно ранней стадии цикла вирусной репликации. В некоторых вариантах осуществления рекомбинантный MVA экспрессирует в клеткахHeLa кодируемый антиген в присутствии 40 мкг/мл AraC на уровне, соответствующем 10, 20 или 50%,или который составляет по меньшей мере 50, 55, 60, 65, 70, 75, 80, 85, 90, 95 или 100% от уровня кодируемого антигена в отсутствие AraC. В предпочтительных вариантах осуществления рекомбинантныйMVA экспрессирует в клетках HeLa кодируемый антиген в присутствии 40 мкг/мл AraC на уровне, который превышает уровень кодируемого антигена, в отсутствие AraC. В различных вариантах осуществления рекомбинантный MVA экспрессирует в 2, 3 или 4 раза более высокие количества кодируемого антигена, чем в случая вектора MVA с использованием промотора pS, осуществляющего экспрессию в клетках HeLa и/или клетках CEF в присутствии 40 мкг/мл AraC. В предпочтительном варианте осуществления гибридный ранний/поздний промотор содержит следующую последовательность: Последовательность позднего промотора ATI показана курсивом, а 5 копий оптимизированного раннего промотора р 7.5 подчеркнуты. Оптимизацию раннего промотора р 7.5 проводили по описанной ранее процедуре (Davison and Moss, Structure of Vaccinia Virus Early Promoters, J. Mol. Biol. 210, 749-769,1989). Элементы оптимизированного промотора pHyb (SEQ ID NO:1) показаны ниже: В других вариантах осуществления гибридный ранний/поздний промотор содержит последовательность, которая по меньшей на 80, 85, 90, 95, 98 или 99% гомологична или идентична нуклеотидной последовательности SEQ ID NO:1 или фрагменту из нуклеотидов 15-41 или нуклеотидов 48-81, нуклеотидов 48-87 или нуклеотидов 48-247 последовательности SEQ ID NO:1. На основе информации о консенсусных последовательностях раннего и позднего промоторов, а также информации о влиянии различных нуклеотидных замен на активность раннего и позднего промотора (Davison and Moss, Structure ofVaccinia Virus Early Promoters, J. Mol. Biol. 210, 749-769, 1989) можно изменить промоторную последовательность SEQ ID NO:1 так, чтобы избежать отрицательного влияния на активность промотора. Нуклеотидные последовательности, которые отличаются от SEQ ID NO:1 по одному или нескольким положениям, но которые, по существу, сохраняют такую же (т.е. в диапазоне примерно 20%) активность раннего и позднего промотора, как и промотор SEQ ID NO:1, входят в область настоящего изобретения. В другом предпочтительном варианте настоящее изобретение относится к промотору, содержащему по меньшей мере 2 элемента нуклеотидной последовательности, обладающие по меньшей мере 80, 85,90, 95, 98, 99 или даже 100% гомологией или идентичностью к нуклеотидам фрагмента 48-81 в последовательности SEQ ID NO:1. В особенно предпочтительном варианте осуществления промотор содержит по меньшей мере 2 предпочтительно по меньшей мере 5 элементов нуклеотидной последовательности,соответствующей фрагменту 48-81 в последовательности SEQ ID NO:1. Предпочтительно промотор содержит по меньшей мере один поздний промоторный элемент, предпочтительно поздний промоторный элемент ATI вируса коровьей оспы. Процент идентичности последовательностей может быть определен при визуальном осмотре и с помощью математических расчетов. Альтернативно, процент идентичности двух последовательностей нуклеиновой кислоты может быть определен путем сравнения информации о последовательности с помощью компьютерной программы GAP, версия 6.0, описанной Devereux et al. (Nucl. Acids Res. 12:387,1984) и доступной от University of Wisconsin Genetics Computer Group (UWGCG. Предпочтительные заданные по умолчанию параметры в программе GAP включают (1) унитарную матрицу для сравнения(которая включает показатель 1 для указания идентичности и показатель 0 для указания на отсутствие идентичности) нуклеотидов и взвешенную матрицу для сравнения (Gribskov and Burgess, Nucl. Acids Res. 14:6745, 1986, описанная Schwartz and Dayhoff, eds., Atlas of Protein Sequence and Structure, NationalBiomedical Research S Foundation, p. 353-358, 1979); (2) штраф в размере 3,0 для каждого пропуска и дополнительный штраф 0,10 для каждого знака в каждом пропуске и (3) отсутствие штрафа за концевые пропуски. Могут также использоваться другие программы, используемые специалистами в данной области для сравнения последовательностей. Настоящее изобретение также относится к дефицитному по репликации рекомбинантному вирусу,как определенно выше, для применения в качестве лекарственного средства или вакцины и к применению дефицитного по репликации рекомбинантного вируса, как определенно выше, для получения лекарственного средства или вакцины. Дефицитный по репликации рекомбинантный вирус по настоящему изобретению вводят в диапазоне концентраций 102-109 или 104-109 TCID50 (инфицирующая доза для культуры ткани)50/мл, предпочтительно в диапазоне концентраций, например, 105-5108 TCID50/мл, более предпочтительно в диапазоне концентраций, например, 106-108 TCID50/мл и наиболее предпочтительно в диапазоне концентраций, например, 107-108 TCID50/мл или по меньшей мере в диапазоне концентраций 2-5107-108, или 2-5108-109, в особенности 108 TCID50/мл. Фактическая концентрация зависит от типа используемого вируса и вида вакцинируемого животного. Предпочтительно для вакцинации человека доза составляет 106-109 TCID50, более предпочтительно 107 или 108 TCID50 и наиболее предпочтительно 108 TCID50 или более, в частности 2 или 2,5-5108 или 109. В случае использования MVA-BN характерная для вакцинации человека доза составляет 5107 TCID50 - 5108 TCID50, в частности составляет такую дозу, как 1, 2 или 2,5108 TCID50, при подкожном введении. Можно индуцировать иммунный ответ при однократном введении дефицитного по репликации рекомбинантного вируса, описанного выше, например MVA, в частности штамма MVA-BN и его производных. Обычно можно использовать дефицитный по репликации рекомбинантный вирус по настоящему изобретению, например MVA, в частности,MVA-BN и его производные в режимах гомологичных первичного/бустерного режимов, например, можно использовать рекомбинантный вирус для первичной вакцинации и для усиления иммунного ответа, полученного при первой иммунизации, вводить тот же или родственный рекомбинантный вирус, чем вирус, использованный при первичной вакцинации. Гомологичное первичное/бустерное введение также является предпочтительным вариантом осуществления настоящего изобретения. Дефицитный по репликации вирус рекомбинантный по настоящему изобретению, например MVA,в частности MVA-BN и его производные, также могут использоваться в гетерологичных режимах первичной/бустерной иммунизации, в рамках которых одну или несколько вакцинаций проводят вирусом,определенным выше, а одну или несколько вакцинаций проводят с использованием другого вида вакцины, например вакцины на основе другого вируса, белковой вакцины или вакцины на основе нуклеиновой кислоты. Способ введения может быть внутривенным, внутримышечным, внутрикожным, интраназальным или подкожным. Предпочтительным способом является внутривенное, внутримышечное или, в частности, подкожное введение. Однако может использоваться любой другой способ введения, такой как скарификация. Настоящее изобретение также относится к фармацевтической композиции или вакцине, содержащей определенный выше дефицитный по репликации рекомбинантный вирус и, необязательно, фармацевтически приемлемый носитель, разбавитель, адъювант и/или добавку. Специалистам в данной области известно множество способов получения вирусных композиций, а также способов их хранения. В контексте настоящего изобретения и, в частности, для получения поксвирусных композиций см. WO 03/053463. Неограничивающие примеры вспомогательных веществ включают воду, физиологический раствор,глицерин, этанол, смачивающие вещества или эмульгаторы, pH-забуферивающие вещества, консерванты, стабилизаторы или т.п. Подходящие носители, как правило, выбирают из группы, включающей крупные медленно метаболизируемые молекулы, такие как, например, белки, полисахариды, полимолочные кислоты, полигликолевые кислоты, полимерные аминокислоты, сополимеры аминокислот, агрегаты липидов или т.п. Для получения вакцин рекомбинантный вирус MVA по настоящему изобретению преобразуют в физиологически приемлемую форму. Подходящие для этой цели препараты зависят от типа вирусов и известны специалистам в данной области. В случае поксвирусных вакцин такое преобразование может быть осуществлено с учетом опыта получения противооспенных вакцин (как описано в работе Stickl, H.et al. Dtsch. med. Wschr. 99, 2386-2392 [1974]). Например, очищенный вирус хранят при температуре-80 С с титром 5108 TCID50/мл в составе композиции в 10 мМ Tris, 140 мМ NaCl pH 7,4. В одном из вариантов осуществления дефицитный по репликации рекомбинантный вирус по настоящему изобретению используют для получения инъецируемых препаратов вакцин. В частности, примерно 102-108 частиц вируса лиофилизируют в 100 мл фосфатно-буферного раствора (ФБР) в присутствии 2% пептона и 1% альбумина человека в ампуле, предпочтительно в стеклянной ампуле. В другом неограничивающем примере инъецируемые препараты вакцин получают путем ступенчатой сушки с замораживанием вируса в композиции. В некоторых вариантах указанная композиция может содержать дополнительные добавки, такие как маннит, декстран, сахар, глицин, лактоза или поливинилпирролидон,или другие вспомогательные вещества, такие как антиоксиданты или инертный газ, стабилизаторы или рекомбинантные белки (например, сывороточный альбумин человека), подходящие для введения in vivo. Затем стеклянную ампулу герметизируют и затем ампулу можно хранить при температуре от 4 С до комнатной температуры в течение нескольких месяцев. Однако если нет необходимости в срочном введении, то ампулу хранят при температуре предпочтительно ниже -20 С. Для вакцинации или терапии, лиофилизат может быть растворен в 0,1-0,5 мл водного раствора,предпочтительно, физиологического или Tris буфера и затем вводят системно или местно, например парентерально, подкожно, внутримышечно, путем скарификации или любым другим способом введения,известным практикующим специалистам в данной области. Способ введения, доза и количество введения могут быть оптимизированы специалистом в данной области с использованием известных способов. Однако чаще всего пациента вакцинируют путем второй инъекции примерно через один месяц - шесть недель после первой инъекции вакцины. Третья, четвертая и последующая инъекции обычно могут быть проведены через 4-12 недель, предпочтительно 4-6 недель после предшествующего введения. В одном варианте млекопитающих, которые включают крыс, кроликов, мышей и людей, иммунизируют путем введения дозировки дефицитного по репликации рекомбинантного вируса, в частностиMVA, указанному индивидууму, предпочтительно, человеку. В одном из вариантов первая дозировка, а также вторая и последующие дозировки (т.е. третья, четвертая, пятая и т.п.), в особенности рекомбинантного MVA, включают 108 TCID50 рекомбинантного вируса. В другом аспекте настоящее изобретение относится к дефицитному по репликации рекомбинантному вирусу, к фармацевтической композиции или вакцине, согласно приведенному выше определению,для индукции T-клеточного ответа в организме хозяина на указанный по меньшей мере один антиген и/или один антигенный эпитоп. Кроме того, настоящее изобретение относится к применению дефицитного по репликации рекомбинантного вируса, фармацевтической композиции или вакцины, согласно приведенному выше определению, для получения лекарственного средства для индукции T-клеточного ответа в организме хозяина на указанный по меньшей мере один антиген и/или один антигенный эпитоп. В предпочтительном варианте указанный T-клеточный ответ представляет собой ответ T-клетокCD8. Иммунизация с использованием дефицитного по репликации рекомбинантного вируса по настоящему изобретению, в частности рекомбинантного MVA, может вызывать сильные ответы T-клеток CD8. В предпочтительных вариантах осуществления после первого первичного и по меньшей мере двух бустерных введений, введение которых проводят с интервалами, составляющими по меньшей мере одну неделю, рекомбинантный MVA вызывает ответ T-клеток CD8 в организме хозяина против колируемого антигена, где указанный ответ выше, чем T-клеточный CD8 ответ против иммунодоминантногоCD8 против антигена, контролируемый оптимизированным гибридным ранним/поздним промотором по настоящему изобретению, усилен, по сравнению с ответом против того же антигена, под контролем синтетического мощного pS промотора или аналогичного раннего/позднего промотора. Предпочтительно после третьего, четвертого, пятого и т.п. введения в организме хозяина проявляется иммунодоминантныйT-клеточный ответ против кодируемого антигена, т.е. T-клеточный эпитоп CD8, полученный из рекомбинантного антигена, превращается в иммунодоминантный эпитоп. Наиболее предпочтительно после третьего, четвертого, пятого и т.п. введения рекомбинантный MVA индуцирует T-клеточный ответ CD8 в организме хозяина против кодируемого антигена, который вовлекает по меньшей мере 10, 15, 20, 25, 30 или 35% от общего числа T-клеток CD8. В предпочтительном варианте T-клеточный ответ CD8 против рекомбинантного ангтигена, индуцируемый после третьего, четвертого, пятого и т.п. введения рекомбинантного MVA, содержащего оптимизированный гибридный ранний/поздний промотор по настоящему изобретению, по меньшей мере на 20% превышает T-клеточный CD8 ответ, получаемый после введения рекомбинантного MVA, содержащегоpS промотор. В других предпочтительных вариантах T-клеточный ответ CD8, получаемый после третьего, четвертого, пятого и т.п. введения, выше на 30, 40, 50, 60, 70, 80, 90%. В наиболее предпочтительном варианте получаемый T-клеточный ответ CD8 выше на 100%. Как используется в настоящем изобретении, под термином "получаемый T-клеточный ответ" следует понимать так, что T-клеточный ответ индуцируется, возникает и/или усиливается. Дефицитный по репликации рекомбинантный вирус по настоящему изобретению может использоваться для лечения большого числа млекопитающих, в том числе человека и даже людей с нарушенным иммунитетом. В предпочтительных вариантах осуществления указанное лечение включает три, четыре, пять или даже больше введений (что соответствует первому примированию с последующим проведением по меньшей мере двух, трех, четырех, пяти или даже более бустерных введений) дефицитного по репликации рекомбинантного вируса, предпочтительно рекомбинантного MVA в организм-хозяина. Введение рекомбинантного вируса проводится в виде первичного-бустерного введения, т.е. проводится по меньшей мере три введения, которые включают первую инокуляцию (первичную инокуляцию/иммунизацию) и затем вторую и третью инокуляцию (бустерные инокуляции/иммунизации). В контексте настоящего изобретения термин "хозяин" обозначает любой вид животного, в частности позвоночное животное. Предпочтительными являются млекопитающие, включающие людей. Другие конкретные примеры подходящих животных включают домашних животных, таких как собаки, кошки,экономически значимых животных, таких как телята, крупный рогатый скот, овцы, козы, лошади, свиньи, а также других животных, таких как мыши, крысы. Для таких видов животных и для человека особенно предпочтительными вирусами являются вирусы MVA и DISC-HSV. Настоящее изобретение может также использоваться применительно к экономически значимым птицам, таким как индейки, утки, гуси и куры, если используются вирусы, которые способны инфицировать клетки птиц, но не способны продуцировать инфекционное вирусное потомство в указанных клетках.T-клеточный ответ на указанный по меньшей мере один антиген и/или один антигенный эпитоп может быть индуцирован в рамках гетерологичных первичного/бустерного режимов, при которых одну или несколько вакцинаций проводят вирусом, определенным выше, и одну или несколько вакцинаций проводят с использованием другого типа вакцины, например вакцины на основе другого вируса, белковой вакцины или вакцины на основе нуклеиновой кислоты. Однако предпочтительно указанныйT-клеточный ответ индуцируется с использованием гомологичных первичного/бустерного режимов, при которых тот же или родственный дефицитный по репликации рекомбинантный вирус используют и для первичной, и для бустерной вакцинации. Соответственно, в другом предпочтительном варианте указанный T-клеточный ответ индуцируется с использованием режима иммунизации, включающего гомологичные первичные/бустерные введения. В других вариантах указанный T-клеточный ответ индуцируется с использованием режима иммунизации, предусматривающего по меньшей мере три или по меньшей мере четыре введения дефицитного по репликации рекомбинантного вируса или фармацевтической композиции или вакцины, согласно приведенному выше описанию. Настоящее изобретение также относится к набору, содержащему по меньшей мере два флакона для первичной/бустерной иммунизации, содержащие указанный дефицитный по репликации рекомбинантный вирус для первой инокуляции ("первичная инокуляция") в первом флаконе/контейнере и, по меньшей мере, для второй, и/или третьей, и/или последующих инокуляций ("бустерная инокуляция") во втором и/или последующих флаконах/контейнерах. Указанный набор может включать по меньшей мере один, два, три, четыре или большее количество контейнеров или флаконов с рекомбинантным вирусом, вместе с инструкциями по введению указанного вируса индивидууму. В предпочтительном варианте указанным индивидуумом является человек. В инструкции может быть указано, что рекомбинантный вирус вводится индивидууму в виде многократных дозировок (например, 2, 3, 4, 5, 6 и т.п.) в определенные временные точки (например, по меньшей мере через 4 недели, по меньшей мере через 6 недель, по меньшей мере через 8 недель после предшествующего введения). Предпочтительно в инструкции указывается, что рекомбинантный вирус вводится по меньшей мере в виде 3 или по меньшей мере в виде 4 дозировок. Если вакцина представляет собой MVA-BN вектор или его производное, содержащее ДНК по настоящему изобретению, то в конкретном варианте настоящее изобретение относится к набору для вакцинации, включающему вирусный вектор MVA-BN по настоящему изобретению для первой вакцинации("примирование") в первом флаконе/контейнере и, по меньшей мере, для второй вакцинации и третьей вакцинации ("бустинг") во втором/третьем/флаконе/контейнере. Краткое описание чертежей Фиг. 1. Последовательность и схема, иллюстрирующая организацию раннего и позднего промоторных элементов в pHyb, р 7.5 и pS.A - Схематическое представление промоторов р 7.5, pS и pHyb. Ранний и поздний промоторные элементы не соотнесены со шкалой.B - Нуклеотидная последовательность промоторов р 7.5 (SEQ ID NO:5), pS (SEQ ID NO:2) и pHyb(SEQ ID NO:1). Область, в которой ранний промоторный элемент был оптимизирован, помещена в рамку в последовательности р 7.5, а оптимизированная последовательность показана ниже. Одиночная жирная линия: поздний промоторный элемент. Двойная линия: ранний промоторный элемент. Фиг. 2. Экспрессия eGFP, осуществляемая рекомбинантными MVA. Рекомбинантные MVA, содержащие открытую рамку считывания eGFP под контролем указанных промоторов, использовали для инфицирования HeLa (А, С) и клеток CEF (В) с множественностью инфекции (MOI или m.o.i.), равной 5. Клетки либо обрабатывали цитозинарабинозидом (+AraC), либо оставляли необработанными (-AraC) в процессе инфицирования (А, В). Клетки собирали через 16 ч после инфицирования (А, В) и анализировали методом проточной цитометрии для оценки экспрессии eGFP. С) Клетки HeLa инфицировали MVA-p7.5-eGFP ("p7.5"), MVA-pS-eGFP ("pS") и MVA-pHyb-eGFP ("pHyb") или инкубировали в среде ("ложный вариант"). В указанные временные точки после инфицирования,клетки собирали и анализировали методом проточной цитометрии для оценки экспрессии eGFP. Эксперименты повторяли независимо по меньшей мере два раза. Фиг. 3. Анализ OVA- и MVA-специфичных ответов T-клеток CD8 на куриный овальбумин, индуцированных рекомбинантными MVA. Использовали MVA-p7.5-OVA, MVA-pS-OVA ("pS") и MVA-pHyb-OVA ("pHyb") для внутрибрюшинной (в/б) иммунизации групп животных, состоявших из 5-6 мышей BALB/c, в дозе 108 TCID50 в расчете на мышь (А). Мышам вводили бустерные инъекции путем второй (В) и третьей (С) в/б инъекции через 4 и 8 недель после первой иммунизации соответственно. Анализировали лейкоциты крови через 68 дней после первой и через 6 дней после второй и третьей иммунизаций для индукцииOVA-специфичных ("OVA") и вектор-специфичных ("B8R") T-клеточных CD8 ответов. Количественную оценку антигенспецифичных T-клеток CD8 проводили путем внутриклеточного окрашивания цитокином, на IFN-, через 6-часовой период рестимуляции и просеивания, на CD4CD8+ или CD19-CD8+ лимфоциты. Лейкоциты иммунизированных животных, инкубированные без пептида, служили в качестве контролей ("без пептида"). Указаны проценты OVA- и BSR-специфичных клеток от общего числаT-клеток CD8 (А-С). Проценты OVA- (CD8OVA) и B8R-специфичных (CD8B8R) T-клеток CD8 использовали для расчета отношения числа клеток CD8OVA к клеткам CD8B8R(D). Показатель log10 полученных соотношений использования для вычисления стандартной ошибки и критерия Стьюдента. Приведены объединенные результаты двух независимых экспериментов (A-D). Фиг. 4. Кинетика OVA- и MVA-специфичных T-клеточных CD8 ответов после трех иммунизаций рекомбинантными MVA. Использовали MVA-p7.5-OVA, MVA-pS-OVA ("pS"), MVA-pHyb-OVA ("pHyb") для внутрибрюшинной (в/б) иммунизации групп животных, состоявших из 5-6 мышей BALB/c, в дозе 108 TCID50 в расчете на мышь (А). Проводили бустинг мышей путем второй (В) и третьей (С) в/б инъекции через 4 и 8 недель после первой иммунизации соответственно. Анализировали лейкоциты крови через 4, 6 и 8 дней после 3-й иммунизации для индукции OVA-специфичных ("OVA") и вектор-специфичных ("B8R")T-клеточных ответов CD8. Количественную оценку антигенспецифичных T-клеток CD8 проводили путем внутриклеточного окрашивания цитокином, на IFN-, после 6-часового периода рестимуляции и просеивания для CD4CD8+ (верхняя панель) или путем окрашивания декстрамером, на MHC, класс I (нижняя панель). Лейкоциты иммунизированных животных, инкубированные без пептида, служили в качестве контролей ("без пептида"). Указаны проценты OVA- и B8R-специфичных клеток от общего числаT-клеток CD8. Фиг. 5. OVA- и MVA-специфичные T-клетки CD8 памяти. Получали лейкоциты мышей BALB/c, иммунизированных MVA-p7.5-OVA, MVA-pS-OVA иMVA-pHyb-OVA, и анализировали через 28 дней (А) и 92 дня (В) после третьей иммунизации ICCS наIFN-, для количественной оценки OVA- и В 8R-специфичных T-клеток CD8. Результаты для животных,исследованных в одном из двух экспериментов, показаны на фиг. 3. Трех из пяти мышей, иммунизированных три раза с использованием MVA-pHyb-OVA, и пять мышей, иммунизированных три раза с использованием MVA-p7.5-OVA и MVA-pS-OVA, вакцинировали четвертый раз соответствующими OVAэкспрессирующими MVA рекомбинантами через 14 недель после третьей иммунизации. Мышей анализировали с использованием IFNICCS на OVA- и В 8R-специфичные T-клетки CD8 в крови (С) и селезенке (D) через 6 дней после бустерной иммунизации. Лейкоциты иммунизированных животных, инкубированные без пептида, служили в качестве контролей ("без пептида"). Примеры Приведенные ниже примеры дополнительно иллюстрируют настоящее изобретение. Для специалистов в данной области очевидно, что приведенные примеры не предназначены для ограничения возможностей использования технологии, описанной в тексте настоящего изобретения и в прилагаемых примерах. Статистический анализ данных проводили с использованием двустороннего дисперсионного анализа (ANOVA), если особо не указано иное. Пример 1. Получение рекомбинантных вариантов MVA-BN. Конструировали гибридный ранний/поздний промотор, обозначенный как pHyb, который содержал поздний элемент ATI вируса коровьей оспы и пять организованных в виде тандема ранних промоторных элементов (фиг. 1). Ранние промоторные элементы были основаны на промоторе р 7.5 и дополнительно были оптимизированы с использованием опубликованных данных (Broyles, S.S. 2003. Vaccinia virusB. Moss. 1989. Structure of vaccinia virus early promoters. J. Mol. Biol. 210:749-769). Промотор pHyb сравнивали с широко используемым синтетическим промотором pS, который обеспечивает высокие уровни генной экспрессии, а также с природным промотором р 7.5 (Cochran, М.А., С. Puckett, and В. Moss. 1985.In vitro mutagenesis of the promoter region for a vaccinia virus gene: evidence for tandem early and late regulatory signals. J. Virol. 54:30-37) (фиг. 1). Указанные промоторные конструкции клонировали против направления считывания гена в открытых рамках считывания для куриного овальбумина (OVA) или eGFP и вводили в геномы MVA вирусов путем гомологичной рекомбинации. Проводили сборку pHyb промотора с использованием промотора, направляющего экспрессию белка включения А-типа (ATI) в вирусе коровьей оспы (Funahashi, S., Т. Sato, and H. Shida. 1988. Cloning andthat directs high levels of expression of cloned genes in mammalian cells. Proc. Natl. Acad. Sci. USA, 85:94319435). Пять организованных в виде тандема ранних элементов получали из промотора р 7.5 и модифицировали по 4 нуклеотидным положениям в пределах А-обогащенной критической ядерной области из 16 нуклеотидов, как было описано ранее (Davison, A.J. and В. Moss. 1989. Structure of vaccinia virus earlypromoters. J. Mol. Biol. 210:749-769). Использованный природный промотор 7.5 состоял из фрагмента ДНК со 104 парами оснований, содержащего поздний и ранний промоторный элемент. Последовательность мощного синтетического ранено/позднего промотора pS содержала 40 нуклеотидов, которые точно подходили последовательности, описанной ранее (Chakrabarti, S., J.R. Sisler, and В. Moss. 1997. Compact,synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques, 23:1094-1097). Рекомбинантные MVA получали с использованием клонированного варианта генома MVA-BN в искусственной бактериальной хромосоме (ВАС). В общих чертах, процедура состояла в том, что pHyb иpS промоторные конструкции клонировали против направления считывания информации в открытых рамках считывания для куриного овальбумина (OVA) или усиленного зеленого флуоресцентного белка(eGFP). Указанные кассеты экспрессии фланкировали гомологичными плечами из примерно 45 нуклеотидов путем ПЦР и вводили в межгенный участок между генами MVA136 и MVA137 по процедуре гомологичной рекомбинации с получением рекомбинантных MVA-BAC. Инфекционные вирусы выделяли из ВАС путем трансфекции ДНК ВАС в клетки ВНК-21 и суперинфекции вирусом фибромы Шоупа, в качестве хелперного вируса. После трех пассажей в клетках CEF получали не содержащие хелперных вирусов (что подтверждали результатами ПЦР) MVA-pHyb-eGFP и MVA-pHyb-OVA, экспрессирующие либо eGFP, либо OVA под контролем промотора pHyb и MVA-pS-eGFP, и MVA-pS-OVA, экспрессирующие либо eGFP, либо OVA под контролем промотора pS. Пример 2. Клеточная культура и остановка клеточного цикла под действием AraC. Получали клетки первичных эмбриональных фибробластов цыпленка (CEF) из 11-дневных эмбрионов и культивировали их в среде VP-SFM (бессывороточная среда; Invitrogen, Karlsruhe, Germany). Клетки HeLa культивировали в DMEM/10% ФСТ (Invitrogen). Клетки инфицировали в дозе 10 TCID50 на клетку рекомбинантами MVA, экспрессирующими eGFP под контролем указанных промоторов. По прошествии указанного периода времени, инфицированные клетки собирали путем обработки трипсином и анализировали методом проточной цитометрии для оценки уровней экспрессии eGFP с использованием прибора для проточной цитометрии LSR II (BD Biosciences, Heidelberg, Germany). Если показано, в среду в ходе инфицирования добавляли цитозинарабинозид (AraC) в конечной концентрации 40 мкг/мл для остановки репликации MVA в ранней фазе. Пример 3. Иммунизация мышей. Самок 6-8-недельных мышей C57BL/6 приобретали от компании Harlan Winkelmann, Германия. Животных иммунизировали в группах по 5 мышей внутрибрюшинной инъекцией при инокуляции 200 мкл, содержащих 108 TCID50 соответствующих рекомбинантов MVA в точках 0, 4, 8 недель, и через 12 недель или 22 недели проводили анализ на T-клетки, а в точках 0, 2, и 4 недели - анализ на наличие анти-OVA и анти-MVA антител. Кровь отбирали через хвостовую вену в указанные временные точки и обрабатывали по описанной ниже процедуре для анализа T-клеточных CD8 ответов. Если показано, отбирали образцы селезенки через 7 дней после последней иммунизации для анализа T-клеточных CD8 ответов. Пример 4. Внутриклеточное окрашивание цитокином (ICCS). У иммунизированных животных отбирали кровь из хвостовой вены и 100-120 мкл крови в расчете на каждую мышь, ресуспендировали в 2 мл ФБР (pH 7,4) содержащего 4% фетальной сыворотки теленка(FCS), 2 мМ этилендиаминтетрауксусной кислоты (ЭДТА) и 2,5 Ед/мл гепарина. Образцы крови разделяли на три аликвоты и проводили лизис эритроцитов с использованием лизирующего буфера для эритроцитов (Red Blood Cell Lysing Buffer (Sigma-Aldrich, Steinheim, Germany. Мононуклеарные клетки периферической крови (РВМС) ресуспендировали в 2 мл среды RPMI/10% ФСТ, содержащей 0,05 мМ-меркаптоэтанола, 1 мкл/мл GolgiPlug (BD Biosciences), блокирующего секрецию цитокинов по пути экзоцетоза, и 1 мкг/мл пептидов OVA257-268 SIINFEKL (SEQ ID NO:3; "OVA"), B8R20-27 TSYKFESV(SEQ ID NO:4; "B8R") или без пептида ("без пептида"). Пептиды приобретали от компании Prolmmune(Oxford, UK). Определяли частоту T-клеток CD8 применительно к иммунодоминантному H-2Kbограниченному эпитопу TSYKFESV, полученному из аминокислотного участка 20-27 раннего белка B8Rdesign and characterization of smallpox vaccines. J. Exp. Med. 201:95-104), в качестве меры, иллюстрирующей ответы вектор-специфичных T-клеточных CD8 ответов. РВМС инкубировали в течение 5 ч при температуре 37 С в атмосфере 5% CO2, собирали центрифугированием, ресуспендировали 3 мл холодного ФБР/10% ФСТ/2 мМ ЭДТА, pH 7,4 и хранили в течение ночи при температуре 4 С. На следующий день РВМС окрашивали анти-CD8 а-Pac-Blue, анти-CD4-PerCP-Cy5.5, анти-CD62L-РЕ-Су 7 антителами, а в некоторых экспериментах - анти-CD127-АРС антителами (все антитела были приобретены от компанииBD Biosciences). РВМС инкубировали с соответствующими разведениями указанных антител в течение 30 мин при температуре 4 С в темноте. После промывки клетки фиксировали и пермеабилизовали с использованием набора Cytofix/Cytoperm Plus (BD Biosciences) в соответствии с инструкциями производителя. После промывки РВМС окрашивали на внутриклеточный интерферон- (IFN-), фактор некроза опухолевой ткани- (TNF-) с использованием FITC-конъюгированного анти-IFN- антитела и РЕ-конъюгированного анти-TNF- антитела (BD Biosciences). Антитела разбавляли в пермеабилизующем/промывочном буфере (BD Biosciences) и РВМС окрашивали в течение 20 мин при температуре 4 С в темноте. После промывки окрашенные клетки анализировали методом проточной цитометрии в системе BD Biosciences LSR II. Пример 5. Окрашивание с использованием пентамера и декстрамера MHC класса I. У иммунизированных животных отбирали кровь из хвостовой вены и 100-120 мкл крови в расчете на каждую мышь, ресуспендировали в 2 мл ФБР (pH 7,4), содержащего 4% фетальной сыворотки теленка(ФСТ), 2 мМ этилендиаминтетрауксусной кислоты (ЭДТА) и 2,5 Ед/мл гепарина. Весь образец или его аликвоту, которые не использовали для ICCS, сразу окрашивали на OVA- и В 8R-специфичные T-клетки(TSYKFESV-декстрамер) и объединяли в одной реакции окрашивания с анти-CD8a-Pac-Blue антителом. После промывки окрашенные клетки анализировали методом проточной цитометрии в системе BDBiosciences LSR II. Пример 6. Метод ELISA для детекции MVA-специфичных антител в мышиной сыворотке. В лунки 96-луночных планшетов вносили неочищенный экстракт MVA-BN-инфицированных клеток CEF. Двукратные серийные разведения сыворотки инкубировали в течение 1 ч при комнатной температуре. При необходимости, делали предварительные разведения мышиных сывороток. Для детекции планшеты инкубировали с антимышиным IgG-HRP антителом овцы для детекции (Serotec) в течение 1 ч при комнатной температуре. ТМБ (Sigma-Aldrich) использовали в качестве субстрата, и реакцию останавливали при добавлении 1 М H2SO4 (Merck). Показатели ОП измеряли при длине волны 450 нм на приборе Tecan F039300 Sunrise Absorbance Reader (Maennedorf, Switzerland). Пример 7. Метод ELISA для детекции OVA-специфичных антител в мышиной сыворотке. В лунки 96-луночных планшетов вносили по 0,25 мкг/лунку куриного овальбумина (Sigma-Aldrich). Двукратные серийные разведения сыворотки инкубировали в течение ночи при температуре 4 С. При необходимости, делали предварительные разведения мышиных сывороток. К лункам планшета добавляли биотинилированный ослиный анти-мышиный IgG H+L (Dianova) на 2 ч при комнатной температуре. Для детекции биотинилированного вторичного антитела к лункам планшета добавляли стрептавидинHRP (Amersham Biosciences) и инкубировали в течение 2 ч при комнатной температуре. Для развития окрашивания использовали ABTS (Sigma-Aldrich)/0,03% Н 2 О 2/0,1 М лимонную кислоту. Показатели ОП измеряли при длине волны 405 нм, стандартная длина волны составляла 492 нм с использованием прибора Тесап F039300 Sunrise Absorbance Reader. Пример 8. Экспрессия eGFP, направляемая рекомбинантными MVA. Экспрессия eGFP в необработанных клетках HeLa, которые были непермиссивными для MVA, была аналогичной для всех трех исследованных промоторов (фиг. 2 А). В необработанных клетках CEF, которые являются пермиссивными для MVA, промоторы pS и р 7.5 осуществляли более высокий уровень экспрессии eGFP, чем промотор pHyb (фиг. 2 В). Для отдельного анализа ранней генной экспрессии цикл инфекции MVA останавливали на ранней фазе путем обработки AraC в течение 16 ч. В этих условиях промотор pHyb приводил к значительно более высокому уровню экспрессии eGFP, чем промоторы pS и р 7.5 (фиг. 2 А, В). Фактически, на уровни eGFP в MVA-pHyb-eGFP-инфицированных клетках не влиялAraC. Это указывает на то, что тандемная организация пяти ранних элементов в pHyb (фиг. 1) отвечает за наблюдаемое повышение экспрессии раннего антигена. Анализ кинетики экспрессии eGFP в клетках HeLa показал, что промотор pHyb направлял экспрессию белка в течение первых 30 мин инфекции, тогда как значительный уровень экспрессии eGFP pS промотором не выявлялся до 90 мин (фиг. 2 С). Более того, промотор pHyb направлял высокие уровни экспрессии eGFP в течение первых 6 ч инфекции (фиг. 2 С). Уровни экспрессии при использовании промоторов р 7.5 и pS достигали значений, которые индуцировал промотор pHyb, только в поздней фазе инфекции, спустя более 6 ч. В этой связи во все временные точки в период времени от 0,5 до 6 ч после инфицирования, экспрессия eGFP промотором pHyb значительно превышала его уровень, достигаемый при использовании промоторов р 7.5 и pS. Важно также то, что активность pHyb промотора проявлялась очень рано, в течение 30 мин после инфицирования, тогда как для активности р 7.5 и pS требовалось в три раза больше времени для индукции выявляемой экспрессии eGFP.T-клеточные CD8 ответы против рекомбинантно экспрессированного OVA под контролем промоторов р 7.5, pS и pHyb определяли у мышей C57BL/6 после одной, двух, трех и четырех иммунизаций с использованием 108 TCID50 рекомбинантного MVA на мышь. OVA-специфичный T-клеточный CD8 ответ определяли при проведении ICCS на IFN- после стимуляции Kb-ограниченным, полученным из OVA пептидом SIINFEKL. Для отслеживания T-клеточного CD8 ответа на MVA вектор количественно оценивали по методу ICCS T-клетки CD8, распознающие иммунодоминантный T-клеточный CD8 эпитоп из поксвирусного раннего белка B8R. Через одну неделю после первой иммунизации, независимо от типа использованного промотора, наблюдались сходные пропорции OVA-специфичных T-клеток CD8(фиг. 3 А). Наблюдалось незначительно более высокое количество T-клеток CD8 после второй иммунизации с использованием MVA-pHyb-OVA, в сравнении с MVA-pS-OVA и MVA p7.5-OVA (фиг. 3 В), которые не были статистически значимыми (р=0,27 и 0,62 соответственно). В 8R-специфичные T-клеточныеCD8 ответы также существенно не различались после первой и второй иммунизаций (р 0,5, фиг. 3 А, В). После третьей иммунизации с использованием MVA-pHyb-OVA наблюдались значительно более сильные OVA-специфичные T-клеточные CD8 ответы, в сравнении с вариантом тройной иммунизацииMVA-pS-OVA (p0,01) и MVA-p7.5-OVA (р 0,001) (фиг. 3 С). Тогда как не отмечалось существенных различий в пропорциях В 8R-специфичных T-клеток CD8 у мышей, иммунизированных с использованием MVA-pHyb-OVA, в сравнении с мышами, которым вводили две других конструкции MVA (р 0,19). Следует отметить, что приведенные результаты демонстрируют возможность существенно повысить число OVA-специфичных T-клеток CD8, даже после двух предшествующих иммунизаций с использованием гомологичных конструкций MVA вируса. Способность MVA вектора вызывать бустинг антигенспецифичных T-клеточных ответов CD8 не зависела от промотора (р 0,001 для OVA-специфичныхT-клеток CD8 после 3, в сравнении с вариантом проведения 2 гомологичных иммунизаций каждой из трех конструкций рекомбинантного MVA). Пропорция OVA-специфичных T-клеток CD8 достигала чрезвычайно высоких значений после трех иммунизаций с использованием MVA-pHyb-OVA. До 20% всех T-клеток CD8 становились OVA-специфичными через 6 дней после третьей иммунизации этой конструкцией (фиг. 3 С). Наблюдались практически равные пропорции OVA-специфичных T-клеток CD8, в сравнении с В 8R-специфичными T-клетками CD8 на 6 день после третьей иммунизацииMVA-pHyb-OVA, по данным ICCS (фиг. 3 С). Частично это было связано со снижением относительного числа В 8R-специфичных T-клеток CD8, указывая предположительно на то, что экспансия примированных OVA-специфичных T-клеток CD8 стимулировалась с высокой эффективностью (фиг. 3 С). Отношение количества OVA-специфичных T-клеток CD8 к количеству В 8R-специфичных T-клеток CD8 существенно отличалось через 3 иммунизации MVA-pHyb-OVA, по сравнению с вариантом использования двух других промоторов (фиг. 3D). Тогда как это соотношение было очень близким для всех трех конструкций после первой иммунизации. Усиливающий эффект pHyb становился заметным после 2 иммунизаций, но не достигал статистической значимости в этой временной точке (фиг. 3D). Взятые вместе, соотношения CD8OVA:CD8B8R указывают на то, что промотор pHyb демонстрирует преимущество в бустерных иммунизациях и особенно после второго бустинга. Следует отметить, что указанные соотношенияCD8OVA:CD8B8R дают основание полагать, что pS промотор был действительно менее эффективным, чем промотор pHyb, но демонстрировал преимущество в сравнении с промотором р 7.5 (фиг. 3D). При анализе OVA-специфичных T-клеточных CD8 ответов в разные временные точки после третьей иммунизации было обнаружено, что OVA-специфичные T-клетки CD8 становились иммунодоминантными в определенные временные точки после бустинга, по данным ICCS (фиг. 4). Реверсирование иммунодоминантности наблюдалось через 4 или 6 дней после третьей иммунизации, в зависимости от эксперимента (фиг. 4 и данные, которые не приведены). В некоторых экспериментах требовались четыре иммунизации MVA-pHyb-OVA для достижения отношения OVA- к B8R-специфичным T-клеткам CD8 1 (данные не приведены). Тогда как после трех или четырех иммунизаций с использованиемMVA-pS-OVA, В 8R-специфичные T-клетки CD8 всегда оставались иммунодоминантными (фиг. 4 и данные, которые не приведены). Эти результаты были подтверждены при использовании метода окрашивания декстамером MHC класса I, который представляет собой модификацию известной методики окрашивания тетрамером MHC класса I. При использовании этой стратегии было выявлено более высокое количество OVA-специфичных, чем B8R-специфичных T-клеток CD8 во все временные точки после третьей иммунизации с использованием MVA-pHyb-OVA, но это никогда не наблюдалось в случае р 7.5 илиpS промотора (фиг. 4). Таким образом, промотор pHyb был способен реверсировать иммунодоминантность T-клеток CD8 в сторону антигена, образуемого под контролем pHyb. Пример 10. T-клетки CD8 памяти. На ранней фазе памяти к 28 дню после третьей иммунизации с использованием MVA-pHyb количество OVA-специфичных T-клеток CD8 еще превышало количество В 8R-специфичных T-клеток CD8 и было значительно выше, чем у мышей, иммунизированных три раза MVA-pS-OVA (фиг. 5 А, р=0,0035). Анализ долговременной памяти T-клеток CD8 в крови мышей через 13 недель после третьей иммунизации показал, что количество OVA-специфичных T-клеток CD8 было все еще значительно выше в вари- 19023832 анте использования MVA-pHyb-OVA, по сравнению с вариантом использования MVA-pS-OVA (фиг. 5 В,р 0,001, по критерию Стьюдента) и MVA-p7.5-OVA (р=0,03). Этот результат был подтвержден при окрашивании пентамерами MHC класса I, позволяющими выявить B8R- и OVA-специфичные T-клеткиCD8 (данные не приведены). Независимо от типа промотора, примерно 70-80% всех OVA-специфичныхT-клеток CD8, выявленных при окрашивании пентамером MHC класса I, относились к фенотипу эффекторной памяти (CD62L-/CD127+) к 12-й неделе после третьей иммунизации (данные не приведены). В заключение следует отметить, что pHyb был способен индуцировать сильный и длительный ответT-клеток CD8 против антигена, и пропорции OVA- и В 8R-специфичных клеток, обнаруженных на ранней фазе после третьей иммунизации конструкциями MVA, по существу, сохранялись в фазе памяти. Пример 11. Эффект четырех иммунизаций. Через 14 недель после третьей иммунизации мышам снова вводили бустерную инъекцию теми же конструкциями MVA для определения, будут ли пропорции OVA-специфичных T-клеток CD8 в крови мышей, иммунизированных MVA-pS-OVA или MVA-p7.5-OVA, возрастать и достигать тех же значений,как и в случае животных, иммунизированных MVA-pHyb-OVA. Однако и в этом случаеMVA-pHyb-OVA был наиболее эффективным и индуцировал наибольшее количествоOVA-специфичных T-клеток CD8 в крови (фиг. 5 С). В отличие от pHyb, pS и р 7.5 не были способны сдвигать характер иммунодоминантности в сторону OVA-специфичных T-клеток CD8 даже после четырех иммунизаций. Анализ спленоцитов из тех же мышей выявил очень близкое значение отношенияOVA-специфичных к В 8R-специфичным T-клеткам CD8, по сравнению с величиной этого отношения для этих двух специализированных вариантов T-клеток CD8 в крови (фиг. 5 С, D), указывая на то, что относительные количества OVA- и В 8R-специфичных T-клеток CD8, полученные при анализе периферической крови, были репрезентативными для всего компартмента T-клеток CD8. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Дефицитный по репликации модифицированный вирус осповакцины Ankara (MVA), кодирующий по меньшей мере один антиген и/или один антигенный эпитоп, где экспрессия указанного антигена и/или эпитопа антигена находится под контролем транскрипционного элемента, содержащего по меньшей мере две копии раннего промоторного элемента, запускающего раннюю экспрессию указанного антигена и/или антигенного эпитопа, и где указанный MVA обладает способностью к репродуктивной репликации in vitro в клетках фибробластов куриных эмбрионов (CEF), но не обладает способностью к репродуктивной репликации в клеточной линии кератиноцитов человека HaCaT, в клеточной линии эмбриональной почки человека 293, в клеточной линии остеосаркомы костей человека 143 В и в клеточной линии цервикальной аденокарциномы человека HeLa. 2. Дефицитный по репликации вирус по п.1, где указанный MVA представляет собой MVA-BN, депонированный в ЕСАСС под номером V00083008. 3. Дефицитный по репликации вирус по п.1 или 2, содержащий предпочтительно по меньшей мере пять копий раннего промоторного элемента. 4. Дефицитный по репликации вирус по любому из предшествующих пунктов, где указанный транскрипционный элемент представляет собой или содержит ранний/поздний промоторный элемент, содержащий поздний элемент, полученный из промотора, отличного от промотора, из которого был получен ранний элемент. 5. Дефицитный по репликации вирус по п.4, где ранний/поздний промоторный элемент представляет собой гибридный ранний/поздний промоторный элемент. 6. Дефицитный по репликации вирус по п.4, где указанный поздний промоторный элемент представляет собой или содержит поздний промоторный элемент ATI вируса коровьей оспы. 7. Дефицитный по репликации вирус по любому из пп.3-6, где указанный ранний промоторный элемент представляет собой или включает ранний промоторный элемент р 7.5. 8. Дефицитный по репликации вирус по п.6 или 7, где указанный ранний/поздний промоторный элемент содержит нуклеотидную последовательность SEQ ID NO:1. 9. Применение дефицитного по репликации вируса по любому из пп.1-8 в качестве лекарственного средства или вакцины для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп. 10. Применение дефицитного по репликации вируса по любому из пп.1-8 для индукцииT-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп. 11. Применение по п.10, где указанный T-клеточный ответ представляет собой ответ CD8 T-клеток. 12. Применение дефицитного по репликации вируса по любому из пп.1-8 для получения лекарственного средства или вакцины для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп. 13. Фармацевтическая композиция, содержащая дефицитный по репликации рекомбинантный вирус по любому из пп.1-8 и фармацевтически приемлемый носитель, разбавитель и/или адъювант. 14. Фармацевтическая композиция по п.13 для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп. 15. Фармацевтическая композиция по п.14, где указанный T-клеточный ответ представляет собой ответ T-клеток CD8. 16. Вакцина, содержащая дефицитный по репликации вирус по любому из пп.1-8 и фармацевтически приемлемый носитель, разбавитель и/или адъювант. 17. Вакцина по п.16 для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп. 18. Вакцина по п.17, где указанный T-клеточный ответ представляет собой ответ CD8 T-клеток. 19. Применение дефицитного по репликации вируса по любому из пп.1-8 при получении лекарственного средства для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп. 20. Применение по п.19, где указанный T-клеточный ответ представляет собой ответ CD8 T-клеток. 21. Применение фармацевтической композиции по п.13 для получения лекарственного средства для индукции T-клеточного ответа в организме хозяина на кодируемый вирусом по меньшей мере один антиген и/или один антигенный эпитоп. 22. Способ индукции T-клеточного ответа в организме хозяина, в том числе человека, отличающийся тем, что указанный способ предусматривает по меньшей мере три или по меньшей мере четыре введения дефицитного по репликации вируса по любому из пп.1-8, фармацевтической композиции по любому из пп.13-15 и/или вакцины по любому из пп.16-18 указанному хозяину. 23. Способ по п.22, где ответ представляет собой CD8 T-клеточный ответ. 24. Набор, включающий по меньшей мере два флакона/контейнера для первичной/бустерной иммунизации, содержащие дефицитный по репликации вирус по любому из пп.1-8, причем первый флакон/контейнер содержит вирус для первичной иммунизации, а другой флакон/контейнер (другие флаконы/контейнеры) содержит(ат) вирус для бустерной иммунизации. 25. Ранний промоторный элемент вируса по пп.1-8, содержащий по меньшей мере два фрагмента нуклеотидной последовательности, соответствующие участку от 48 до 81 нуклеотида SEQ ID NO:1, или по меньшей мере два фрагмента нуклеотидной последовательности, которые по меньшей мере на 80% идентичны участку от 48 до 81 нуклеотида в SEQ ID NO:1. 26. Ранний промоторный элемент по п.25, дополнительно содержащий по меньшей мере один поздний промоторный элемент. 27. Ранний промоторный элемент по п.26, где промотор представляет собой поздний промоторный элемент ATI.
МПК / Метки
МПК: C12N 15/863, C07K 14/07, A61K 39/285
Метки: вирус, репликации, модифицированный, промоторный, элемент, осповакцины, дефицитный, примененние, содержащий
Код ссылки
<a href="https://eas.patents.su/28-23832-deficitnyjj-po-replikacii-modificirovannyjj-virus-ospovakciny-soderzhashhijj-rannijj-pozdnijj-promotornyjj-element-i-ego-primenennie.html" rel="bookmark" title="База патентов Евразийского Союза">Дефицитный по репликации модифицированный вирус осповакцины, содержащий ранний/поздний промоторный элемент, и его примененние</a>
Рекомбинантный модифицированный вирус осповакцины ankara, содержащий ati-промотор вируса коровьей оспы, и способы применения вируса и промотора

Номер патента: 9525
Опубликовано: 28.02.2008
Авторы: Лейрер Соня, Хаули Пол
МПК: A61P 43/00, C12N 15/86
Метки: применения, содержащий, рекомбинантный, осповакцины, промотора, коровьей, оспы, вируса, ati-промотор, анкара, способы, вирус, модифицированный
Формула / Реферат:
1. Рекомбинантный модифицированный вирус осповакцины Ankara (MVA), содержащий в своем геноме экспрессионную кассету, содержащую ATI-промотор вируса коровьей оспы или его производное, и кодирующую последовательность, где экспрессия кодирующей последовательности находится под контролем указанного промотора. 2. Рекомбинантный MVA по п.1, в котором ATI-промотор имеет последовательность SEQ ID NO:1. 3. Рекомбинантный MVA по п.1, в котором производное...
Рекомбинантный модифицированный вирус осповакцины анкара (mva), экспрессирующий гомологичные последовательности, встроенные в поксвирусный геном, и его применение

Номер патента: 20230
Опубликовано: 30.09.2014
Авторы: Хаули Пол, Лейрер Соня
МПК: C12N 5/10, A61K 39/12, C12N 15/39...
Метки: последовательности, mva, встроенные, применение, осповакцины, поксвирусный, гомологичные, рекомбинантный, анкара, экспрессирующий, геном, модифицированный, вирус
Формула / Реферат:
1. Рекомбинантный модифицированный вирус осповакцины Анкара (MVA), используемый для воздействия на иммунный ответ, предпочтительно на его индуцирование у животного, включая человека, содержащий по меньшей мере две гомологичные между собой чужеродные последовательности, которые получены из разных вариантов микроорганизмов, которые не являются чужеродными генами, с гомологией, составляющей по меньшей мере 50%, в котором каждая из указанных...
Рекомбинантный вирус сендай с ослабленной способностью к репликации, предназначенный для использования в качестве вакцин

Номер патента: 15013
Опубликовано: 29.04.2011
Авторы: Шлехт Забине, Боссов Саша, Нойберт Вольфганг Й.
МПК: A61K 48/00, C07K 14/115, C12N 15/86...
Метки: сендай, способностью, вакцин, предназначенный, ослабленной, рекомбинантный, качестве, использования, вирус, репликации
Формула / Реферат:
1. Рекомбинантный вирус Сендай, содержащий вирусный геном с делецией аминокислот 2-77 белка, кодируемого Р-геном, приводящей к потере способности к репликации без потери вторичной способности к транскрипции.2. Рекомбинантный вирус Сендай по п.1, отличающийся тем, что вирусный геном дополнительно содержит по меньшей мере одну последовательность, кодирующую чужеродный генный продукт.3. Рекомбинантный вирус Сендай по п.2, отличающийся тем, что...
Рекомбинантный вирус, содержащий ген теломелизина-gfp

Номер патента: 11880
Опубликовано: 30.06.2009
Авторы: Кио Сатору, Мизугучи Хиройуку, Танака Нориака, Хаякава Такао, Фудживара Тошийоши
МПК: A61K 48/00, C12N 15/09, A61P 35/00...
Метки: рекомбинантный, вирус, ген, теломелизина-gfp, содержащий
Формула / Реферат:
1. Реагент для выявления раковых клеток, отличающийся тем, что он включает рекомбинантный вирус, в котором репликационная кассета, включающая промотор из теломеразы человека, ген Е1А, последовательность IRES и ген Е1В в таком порядке, интегрирована в область Е1 генома вируса и маркировочная кассета, включающая ген, кодирующий маркировочный белок, и промотор, способный регулировать экспрессию гена, кодирующего маркировочный белок, интегрирована в...
Промотор для экспрессии генов в вирусах осповакцины и/или в клетках, инфицированных вирусом осповакцины, и способы его применения

Номер патента: 12846
Опубликовано: 30.12.2009
Автор: Лейрер Соня
МПК: A61K 39/275, C12N 15/86
Метки: вирусом, генов, промотор, применения, способы, клетках, вирусах, инфицированных, экспрессии, осповакцины
Формула / Реферат:
1. Промотор, состоящий из нуклеотидной последовательности или содержащий нуклеотидную последовательность, выбранную из группы, включающей: (i) любую нуклеотидную последовательность SEQ ID NO: 2-4, 6 и 8-12, (ii) нуклеотидную последовательность длиной по меньшей мере 15 нуклеотидов любой последовательности SEQ ID NO: 2-4 и 6 и (iii) производные любой последовательности SEQ ID NO: 8-12, причем в производном не более 10 нуклеотидов являются...
Предыдущий патент: Способ получения состава, содержащего аморфный гидрат окиси железа
Следующий патент: Применение ингибиторов сукцинатдегидрогеназы для контроля sclerotinia ssp.
Случайный патент: Способ очистки водного экстракта растений