4-((феноксиалкил)тио)феноксиуксусные кислоты и аналоги
Номер патента: 13403
Опубликовано: 30.04.2010
Авторы: Ван Айхуа, Демарест Кит Т., Пелтон Патрисия, Чжан Жуй, Диэнджелис Алан






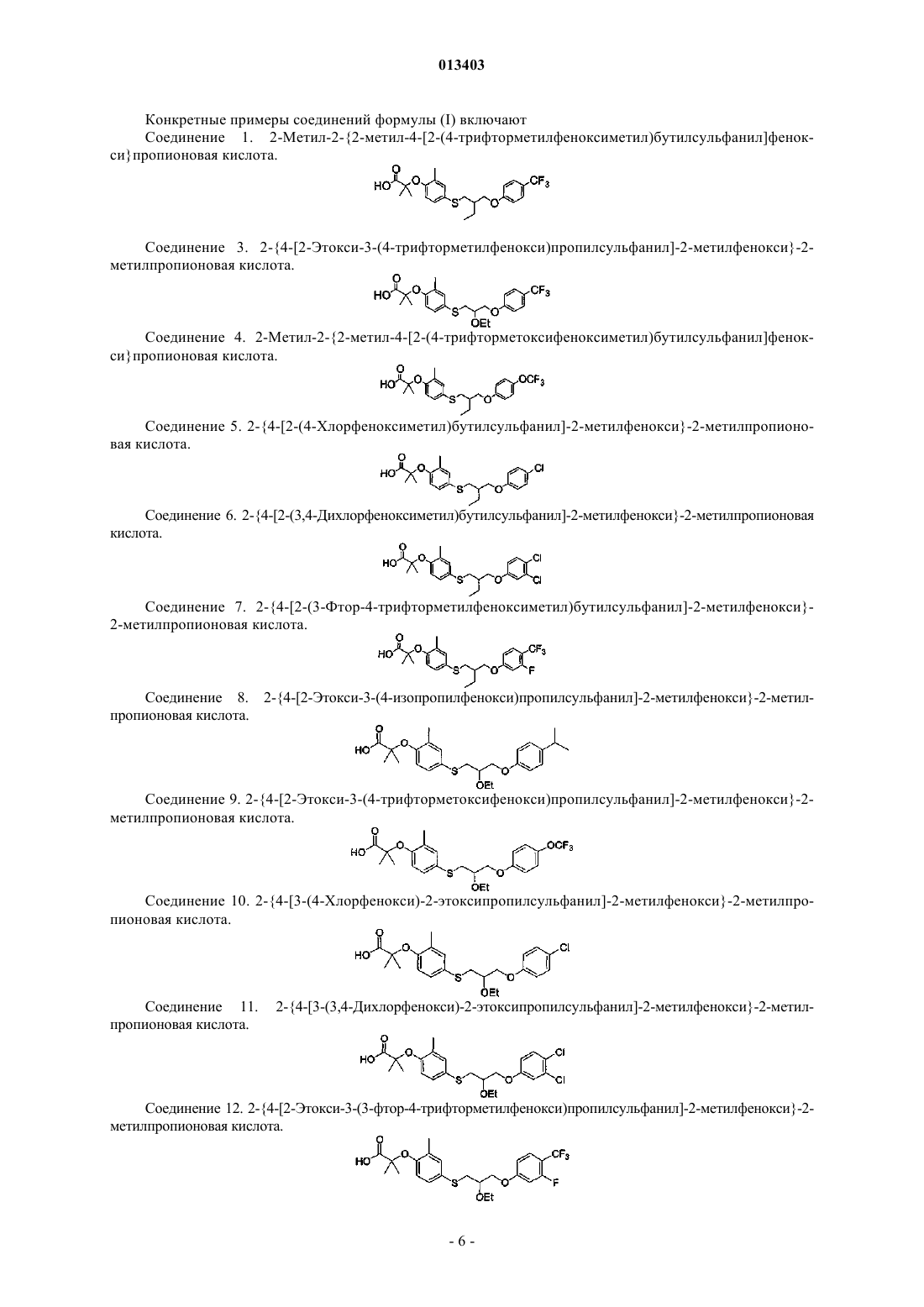



















Формула / Реферат
1. Соединение формулы (I)

где X выбирают из ковалентной связи, S или О;
Y означает S или О;
![]() представляет собой группу, выбираемую из =СН-, -СН=, -СН2-, -СН2-СН2-, =СН-СН2-,
представляет собой группу, выбираемую из =СН-, -СН=, -СН2-, -СН2-СН2-, =СН-СН2-,
-СН2-СН=, =СН-СН= и -СН=СН-;
Z выбирают из О, СН и СН2, при условии, что когда Y означает О, Z означает О;
R1 и R2независимо выбирают из Н, C1-3-алкила, C1-3-алкокси и галогена;
R3 представляет собой Н или галоген;
R4 независимо выбирают из Н, галогена, циано, гидрокси, ацетила, C1-5-алкила, C1-5-алкила, замещенного галогеном, С1-4-алкокси и C1-4-алкокси, замещенного галогеном, при условии, что R3 и R4оба не означают Н;
R5 и R6независимо выбирают из Н, C1-8-алкила и замещенного C1-8-алкила, при условии, что R5и R6 оба не означают Н;
R7 выбирают из галогена, C1-9-алкила, цианоС1-9-алкила, C1-8-алкокси, C2-9-алкенила, C2-9-алкенилокси, C2-9-алкинила, C2-9-алкинилокси, C3-7-циклоалкила, C3-7-циклоалкокси, C3-7-циклоалкил-C1-7-алкила, С3-7-циклоалкил-С1-7-алкокси, C3-7-циклоалкилокси-C1-6-алкила, C1-6-алкокси-С1-6-алкила, С1-5-алкокси-С1-5-алкокси или C3-7-циклоалкилокси-С1-7-алкокси;
R8 означает Н, когда ![]() представляет собой группу, выбираемую из -СН=, -СН2-, -СН2-СН2-, -СН2-СН= и -СН=СН-; или R8 отсутствует, когда
представляет собой группу, выбираемую из -СН=, -СН2-, -СН2-СН2-, -СН2-СН= и -СН=СН-; или R8 отсутствует, когда ![]() представляет собой группу, выбираемую из =СН-, =СН-СН2- и =СН-СН=; и
представляет собой группу, выбираемую из =СН-, =СН-СН2- и =СН-СН=; и
n означает 1 или 2;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где X означает S или О.
3. Соединение по п.1, где X означает ковалентную связь.
4. Соединение по п.2, где X означает О.
5. Соединение по п.1, где Y означает О.
6. Соединение по п.1, где Y означает S.
7. Соединение по п.1, где Z означает О.
8. Соединение по п.1, где Z означает СН или СН2.
9. Соединение по п.1, где ![]() представляет собой группу -СН2- или -СН2-СН2-.
представляет собой группу -СН2- или -СН2-СН2-.
10. Соединение по п.9, где ![]() представляет собой группу -СН2-.
представляет собой группу -СН2-.
11. Соединение по п.1, где ![]() представляет собой группу =СН-, -СН=, =СН-СН2-, -СН2-СН=, =СН-СН= или -СН=СН-.
представляет собой группу =СН-, -СН=, =СН-СН2-, -СН2-СН=, =СН-СН= или -СН=СН-.
12. Соединение по п.1, где R4 выбирают из галогена, C1-4-алкила, C1-4-алкила, замещенного галогеном, C1-3-алкокси и C1-3-алкокси, замещенного галогеном.
13. Соединение по п.1, где R1 и R2независимо выбирают из Н, C1-3-алкила, C1-3-алкокси, F, Cl и Br.
14. Соединение по п.13, где R1 и R2независимо выбирают из Н, метила, метокси, F и Cl.
15. Соединение по п.1, где R4 выбирают из C1-4-алкила, замещенного галогеном, и C1-3-алкокси, замещенного галогеном.
16. Соединение по п.12, где R3 независимо выбирают из Н, F и Cl.
17. Соединение по п.12, где R4 независимо выбирают из трифторметила, фторметила, дифторметила, хлордифторметила, дихлорфторметила, фторметокси, дифторметокси, хлордифторметокси, дихлорфторметокси и трифторметокси.
18. Соединение по п.1, где R3 выбирают из Н, Cl, Br и I и R4выбирают из Н, Cl и метила.
19. Соединение по п.1, где R7 выбирают из C1-7-алкила, C1-6-алкокси, С2-7-алкенила, С2-7-алкенилокси, С2-7-алкинила, С2-7-алкинилокси, C3-7-циклоалкила, C3-7-циклоалкокси, C1-6-алкокси-C1-6-алкила, С1-5-алкокси-С1-5-алкокси и C3-7-циклоалкилокси-С1-7-алкокси.
20. Соединение по п.1, где R7 выбирают из С3-7-циклоалкил-C1-7-алкила, C3-7-циклоалкил-С1-7-алкокси и C3-7-циклоалкилокси-С1-6-алкила.
21. Соединение по п.1, где R8означает Н.
22. Соединение по п.1, где R3 выбирают из Н, F и Cl и R4 выбирают из F, Cl, метила, фторметила, дифторметила, фторметокси, дифторметокси, трифторметила, трифторметокси и метокси.
23. Соединение по п.1, где R1 выбирают из Н, метила, Cl и метокси и R2выбирают из Н, Cl и метила.
24. Соединение по п.23, где X означает ковалентную связь.
25. Соединение по п.23, где X означает ковалентную связь, Y означает S и Z означает О.
26. Соединение по п.1, где X означает О и Y означает О.
27. Соединение по п.1, где X означает О и Y означает S.
28. Соединение по п.1, где Y означает О и Z означает О.
29. Соединение по п.1, где Y означает S и Z означает О.
30. Соединение по п.1, где R8 означает Н и R7выбирают из C1-7-алкила, C1-6-алкокси, С2-7-алкенила, С2-7-алкенилокси, C1-6-алкокси-С1-6-алкила и С1-5-алкокси-С1-5-алкокси.
31. Соединение по п.30, где R7 выбирают из C1-5-алкила, C1-4-алкокси, C2-5-алкенила, C2-5-алкенилокси и С1-5-алкокси-С1-5-алкокси.
32. Соединение по п.30, где R7 выбирают из C1-3-алкила, C1-3-алкокси, C2-4-алкенила, С2-4-алкенилокси и C1-3-алкокси-C1-3-алкокси.
33. Соединение по п.30, где R7 выбирают из метокси, этокси, пропокси, изопропокси, пропенилокси, изопропенилокси, этоксиметокси, метоксиметокси, метоксиметила, метоксиэтила, этоксиметила и этоксиэтила.
34. Соединение по п.1, где R5 и R6независимо означают C1-4-алкил.
35. Соединение по п.34, где R5 и R6означают метил.
36. Соединение по п.1, где
R1 выбирают из Н, метила, Cl и метокси;
R2 выбирают из Н, Cl и метила;
R3 выбирают из Н, F и Cl и
R4 выбирают из F, Cl, метила, трифторметила, трифторметокси, фторметила, фторметокси, дифторметила, дифторметокси и метокси.
37. Соединение по п.1, где
X означает О;
Y означает О;
R3 выбирают из Н, F и Cl и
R4 выбирают из F, Cl, метила, CF3, OCF3 и метокси.
38. Соединение по п.1, где
X означает О;
Y означает S;
R3 выбирают из Н, F и Cl и
R4 выбирают из F, Cl, метила, CF3, OCF3 и метокси.
39. Соединение по п.1, где
X означает ковалентную связь;
Y означает S;
R3 выбирают из Н, F и Cl и
R4 выбирают из F, Cl, метила, CF3, OCF3 и метокси.
40. Соединение по п.1, где
Y означает О;
Z означает О;
R3 выбирают из Н, F и Cl и
R4 выбирают из F, Cl, метила, CF3, OCF3 и метокси.
41. Соединение по п.1, где
Y означает S;
Z означает О;
R3 выбирают из Н, F и Cl и
R4 выбирают из F, Cl, метила, CF3, OCF3 и метокси.
42. Соединение по п.1, где
R3 выбирают из Н, F и Cl;
R4 выбирают из F, Cl, метила, CF3, OCF3 и метокси;
R7 выбирают из C1-7-алкила, C1-6-алкокси, С2-7-алкенила, С2-7-алкенилокси, C1-6-алкокси-C1-6-алкила и С1-5-алкокси-С1-5-алкокси и
R8 означает Н.
43. Соединение по п.1, где
X означает О;
Y означает О;
R7 выбирают из C1-3-алкила, C1-3-алкокси, С2-4-алкенила, С2-4-алкенилокси и C1-3-алкокси-C1-3-алкокси и
R8 означает Н.
44. Соединение по п.1, где
X означает О;
Y означает S;
R7 выбирают из C1-3-алкила, C1-3-алкокси, С2-4-алкенила, С2-4-алкенилокси и C1-3-алкокси-C1-3-алкокси и
R8 означает Н.
45. Соединение по п.1, где
X означает О;
Y означает О;
R1 выбирают из Н, метила, Cl и метокси;
R2 выбирают из Н, Cl и метила;
R3 выбирают из Н, F и Cl;
R4 выбирают из F, Cl, метила, CF3, OCF3 и метокси и
n означает 1.
46. Соединение по п.1, где
X означает О;
Y означает S;
R1 выбирают из Н, метила и Cl;
R2 выбирают из Н, Cl и метила;
R3 выбирают из Н, F и Cl и
R4 выбирают из F, Cl, метила, CF3, OCF3 и метокси.
47. Соединение по п.44, где n=1.
48. Соединение по п.45, где R5 означает C1-3-алкил и R8 означает Н.
49. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически приемлемый носитель.
50. Способ лечения или ингибирования развития опосредованного дельта-PPAR состояния, выбираемого из группы, состоящей из диабета, нефропатии, невропатии, ретинопатии, синдрома поликистоза яичника, гипертензии, ишемии, удара, синдрома раздраженной толстой кишки, воспаления, катаракты, сердечно-сосудистых заболеваний, метаболического синдрома X, гипер-LDL-холестеринемии, гипертриглицеридемии, гиперхолестеринемии, смешанной гиперлипидемии, гипо-HDL-холестеринемии, атеросклероза, ожирения и их осложнений, включающий введение пациенту, нуждающемуся в лечении, фармацевтически эффективного количества композиции по п.49.
51. Соединение, выбранное из группы, состоящей из

или его фармацевтически приемлемая соль.
52. Соединение, выбранное из группы, состоящей из

или его фармацевтически приемлемая соль.
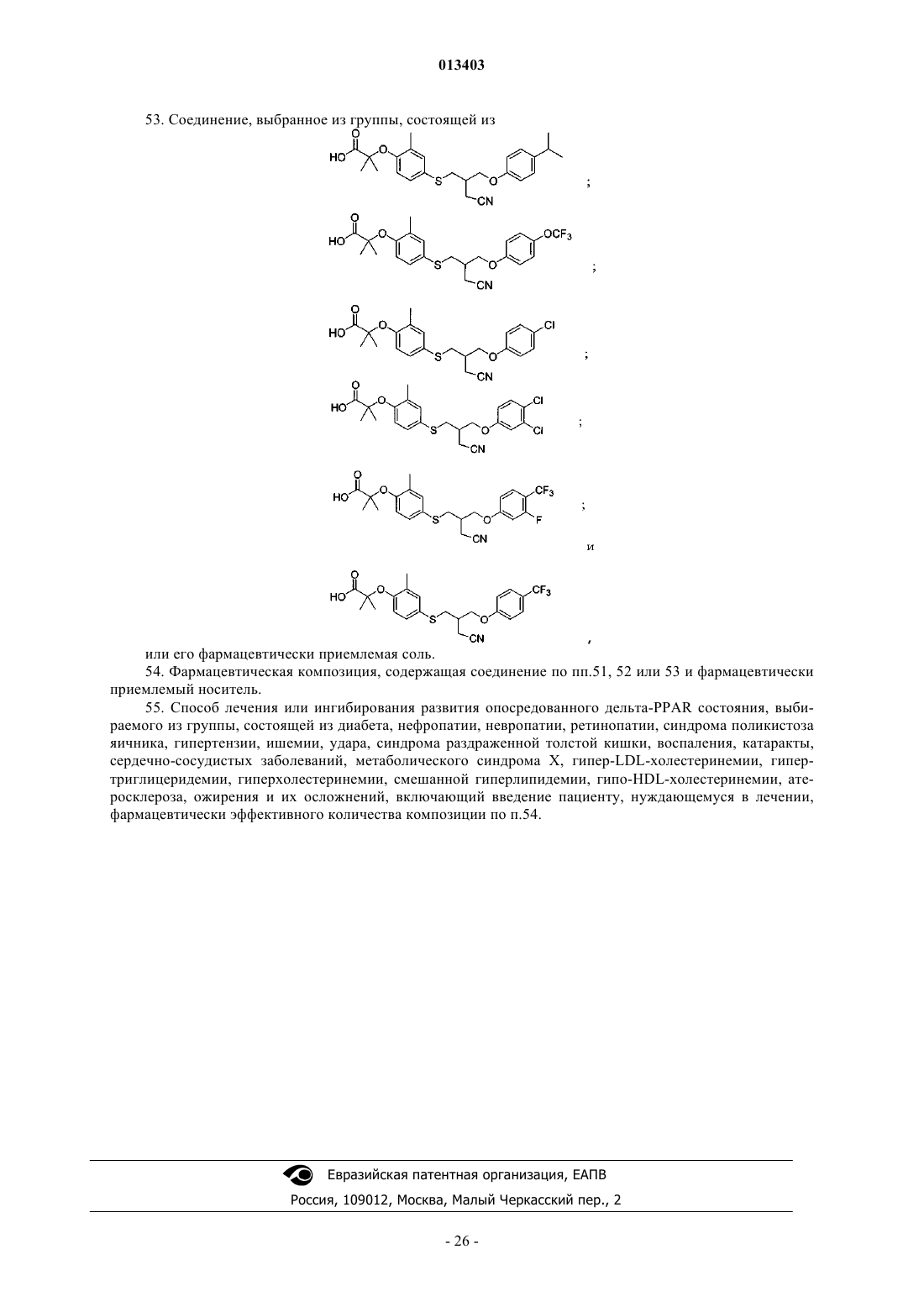
53. Соединение, выбранное из группы, состоящей из


или его фармацевтически приемлемая соль.
54. Фармацевтическая композиция, содержащая соединение по пп.51, 52 или 53 и фармацевтически приемлемый носитель.
55. Способ лечения или ингибирования развития опосредованного дельта-PPAR состояния, выбираемого из группы, состоящей из диабета, нефропатии, невропатии, ретинопатии, синдрома поликистоза яичника, гипертензии, ишемии, удара, синдрома раздраженной толстой кишки, воспаления, катаракты, сердечно-сосудистых заболеваний, метаболического синдрома X, гипер-LDL-холестеринемии, гипертриглицеридемии, гиперхолестеринемии, смешанной гиперлипидемии, гипо-HDL-холестеринемии, атеросклероза, ожирения и их осложнений, включающий введение пациенту, нуждающемуся в лечении, фармацевтически эффективного количества композиции по п.54.
Текст
013403 Перекрестная ссылка на родственные заявки Данная заявка претендует на приоритет предварительной заявки на патент США под номером 60/609942, поданной 15 сентября 2004 года, которая включена во всей ее полноте в данное описание путем ссылки. Область техники, к которой относится изобретение Данное изобретение относится к 4-феноксиалкил)тио)феноксиуксусным кислотам и аналогам, содержащим их композициям и способам их применения. Уровень техники Рецепторы, активируемые пролифераторами пероксисом, (PPAR), являются метаболическими сенсорами, регулирующими экспрессию генов, принимающих активное участие в глюкозном и липидном гомеостазе. Агонисты -подтипа PPAR, такие как LOPID (гемфиброзил) и TRICOR (фенофибрат), и агонисты -подтипа PPAR, такие как AVANDIA (розиглитазонмалеат), применяют для лечения дислипидемии и диабета соответственно. Сообщалось, что другой член этого семейства ядерных рецепторов,активируемый пролифераторами пероксисом дельта-рецептор (PPAR-дельта или PPAR), также является необходимым фактором транскрипции, участвующим в регуляции генов, задействованных в липидном метаболизме и потреблении энергии. Показано, что PPAR-дельта действует как рецептор мембранного канала, модулирующий экспрессию других PPAR (Shi и др., 2002, Proc. Natl. Acad. Sci., США, 99(5): 2613-2618). Каждый подтип рецептора распространен в определенной ткани: 1) PPAR наиболее активно экспрессируется в печени; 2) PPAR обнаружен, главным образом, в жировой ткани; 3) PPAR имеет наиболее широкое распространение повсеместно у взрослой крысы (Braissant и др., 1996, Endocrinology,137(1): 354-366) и во всех протестированных на сегодняшний день тканях человека, включая печень,почки, брюшной жир и скелетные мышцы (Auboeuf и др., 1997, Diabetes, 46(8): 319-1327). Альфа-рецептор, активируемый пролифератором пероксисом (PPAR-альфа или PPAR), является необходимым фактором транскрипции, регулирующим гены, связанные с метаболизмом жирных кислот и действием инсулина. Гены, регулируемые PPAR-альфа, включают ферменты, вовлеченные в бетаокисление жирных кислот, белок транспорта жирных кислот печени и аро-А 1, важный компонент липопротеинов высокой плотности (HDL). Селективная высокая аффинность агонистов PPAR-альфа увеличивает окисление жирных кислот печени, что, в свою очередь, снижает циркуляцию триглицеридов и свободных жирных кислот. Примеры известных агонистов PPAR-альфа, в различной степени пригодных в случае гиперлипидемии, диабета или атеросклероза, включают фибраты, такие как фенофибрат (Fournier), гемфиброзил(Parke-Davis/Pfizer, Mylan, Watson), клофибрат (Wyeth-Ayerst, Novopharm), безафибрат и ципрофибрат, и уреидофибраты, такие как GW 7647, 9578 и 9820 (GlaxoSmithKline). Диабет является заболеванием, которое вызывают или которому способствуют многочисленные факторы, и характеризуется гипергликемией, которая может быть связана с повышенной и преждевременной смертностью вследствие увеличения риска микрососудистых и макрососудистых заболеваний,таких как нефропатия, невропатия, ретинопатия, атеросклероз, синдром поликистоза яичника (PCOS),гипертензия, ишемия, удар и заболевания сердца. Сахарный диабет типа I (IDDM) возникает в результате генетически обусловленной недостаточности инсулина, гормона, регулирующего глюкозный метаболизм. Диабет типа II известен как инсулиннезависимый сахарный диабет (NIDDM) и обусловлен полной резистентностью к инсулину, регулирующему глюкозный и липидный метаболизм в основных, чувствительных к инсулину, тканях, то есть мышцах, печени и жировой ткани. Эта резистентность к инсулину или пониженная чувствительность к инсулину в результате приводит к недостаточной инсулиновой активации поглощения, окисления и аккумуляции в мышцах глюкозы и аномальной инсулиновой репрессии липолиза в жировой ткани, а также к продуцированию и секреции глюкозы в печени. Множество больных, страдающих сахарным диабетом типа II, также страдают ожирением, и полагают, что ожирение вызывает и/или обостряет многие проблемы здоровья и социальные проблемы, такие как ишемическая болезнь сердца, удар, обструктивные приступы апноэ во сне, подагра, гиперлипидемия, остеоартрит, пониженная фертильность и ослабленная психосоциальная функция. Предполагают, что такой класс соединений, как тиазолидиндионы (глитазоны), обладает способностью уменьшать интенсивность многих симптомов NIDDM посредством связывания с рецептором, активируемым пролифераторами пероксисом (PPAR), из семейства рецепторов. Эти соединения увеличивают чувствительность к инсулину в мышцах, печени и жировой ткани в случае отдельных животных моделейNIDDM, приводя к корректировке повышенных уровней в плазме глюкозы, триглицеридов и неэтерифицированных свободных жирных кислот, без появления гипогликемии. Однако у исследуемых животных и/или людей возникают нежелательные эффекты, включая гипертрофию сердца, разжижение крови и токсичность печени. Многие агонисты PPAR, обнаруженные в настоящее время, имеют тиазолидиндионовый цикл в качестве общего структурного элемента. Показано, что агонисты PPAR в высшей степени пригодны для лечения NIDDM и других нарушений, вызываемых резистентностью к инсулину. Троглитазон, розиглитазон и пиоглитазон разрешены для лечения диабета типа II в США. Также имеется ряд указаний на то,-1 013403 что содержащие бензимидазол тиазолидиндионовые производные можно применять для лечения синдрома раздраженной толстой кишки (IBD), воспаления и катаракты (JP 10195057). Сердечно-сосудистые заболевания (CVD) распространены во всем мире и часто связаны с другими болезненными состояниями, такими как диабет и ожирение. В случае многих демографических исследований делались попытки определить факторы риска для возникновения CVD; при этом считают, что к числу наиболее важных факторов относятся высокие уровни в плазме низкой плотности липопротеинхолестерина (LDL-C), высокие уровни в плазме триглицеридов (200 мг/дл) и низкие уровни высокой плотности липопротеин-холестерина (HDL-C). В настоящее время существует немного терапевтических способов, нацеленных на низкие уровни HDL-C и триглицериды. В последнее время опубликованы данные в отношении эффективных лигандов для PPAR, обеспечивающие лучшее понимание их функции, в липидном метаболизме. Основным эффектом этих соединений в случае db/db-мышей (Leibowitz и др., 2000, FEBS, Lett., 473(3): 333-336) и страдающих ожирением макак-резусов (Oliver и др., 2001, Proc. Natl. Acad. Sci., США, 98(9): 5306-5311) является повышение уровня высокой плотности липопротеин-холестерина (HDL-C) и снижение количества триглицеридов с незначительным эффектом в отношении уровня глюкозы (несмотря на то, что у макак снижаются уровни инсулина). HDL-C удаляет холестерин из периферических клеток за счет процесса, называемого обратным транспортом холестерина. Первая и лимитирующая скорость стадия, перенос клеточного холестерина и фосфолипидов к аполипопротеиновому A-I компоненту HDL, опосредуется транспортером АТФ-связывающей кассеты A1 (ABCA1) (Lawn и др., 1999, J. Clin. Investigation, 104(8): R25-R31). Показано, что активация PPAR способствует повышению уровня HDL-C за счет транскрипционной регуляции АВСА 1 (Oliver и др., 2001, Proc. Natl. Acad. Sci., США, 98(9): 5306-5311). За счет индукции экспрессии мРНК АВСА 1 в макрофагах агонисты PPAR могут повышать уровни HDL-C у пациентов и удалять избыток холестерина из нагруженных липидами макрофагов, ингибируя, таким образом, развитие атеросклеротических патологических изменений. Существующая терапия гиперхолестеринемии включает статиновые лекарственные средства, которые снижают уровень LDL-С, но показывают небольшой эффект в отношении HDL-C, и фибраты, агонисты PPAR, которые имеют низкую эффективность и вызывают только умеренное повышение уровня HDL-C. Кроме того, подобно фибратам агонисты PPAR также могут снижать количество триглицеридов, дополнительного фактора риска для сердечно-сосудистых заболеваний и диабета. Показано, что повышенный уровень свободных жирных кислот способствует резистентности к инсулину и развитию диабета (Boden, G. Proceedings of the Associanion of AmericanPhysicians (1999, май-июнь), 111(3), 241-8). Примеры известных агонистов дельта-PPAR, в различной степени пригодных в случае гиперлипидемии, диабета или атеросклероза, включают L-165041 (Leibowitz и др., 2000) и GW501516 (Oliver и др.,Proceedings of the National Academy of Sciences of the United States of America (2001), 98(9), 5306-5311). Обработка дифференцированных моноцитов ТНР-1 с помощью GW501516 индуцирует экспрессию мРНК АВСА 1 и увеличивает отток холестерина из этих клеток. Краткое изложение сущности изобретения Данное изобретение относится к соединениям нижеприводимой формулы (I)X выбирают из ковалентной связи, S или О;R1 и R2 независимо выбирают из Н, C1-3-алкила, C1-3-алкокси и галогена;R4 независимо выбирают из Н, галогена, циано, гидрокси, ацетила, C1-5-алкила, C1-5-алкила, замещенного галогеном; C1-4-алкокси и C1-4-алкокси, замещенного галогеном, при условии, что R3 и R4 оба не означают Н;R5 и R6 независимо выбирают из Н, C1-8-алкила и замещенного C1-8-алкила, при условии, что R5 и R6 оба не означают Н;R8 означает Н, когда представляет собой группу, выбираемую из -СН=, -СН 2-,представляет собой груп-СН 2-СН 2-, -СН 2-СН= и -СН=СН-, или R8 отсутствует, когда пу, выбираемую из =СН-, =СН-СН 2- и =СН-СН=; иn означает 1 или 2; или их фармацевтически приемлемой соли. Данное изобретение также относится к фармацевтическим композициям, содержащим соединение формулы (I) и фармацевтический носитель. Другой аспект согласно данному изобретению включает способы лечения или ингибирования развития состояния, опосредованного дельта-PPAR. Вышеуказанные состояния включают, но не ограничиваясь этим, диабет, нефропатию, невропатию, ретинопатию, синдром поликистоза яичника, гипертензию,ишемию, удар, синдром раздраженной толстой кишки, воспаление, катаракту, сердечно-сосудистые заболевания, метаболический синдром X, гипер-LDL-холестеринемию, дислипидемию (включая гипертриглицеридемию, гиперхолестеринемию, смешанную гиперлипидемию и гипо-HDL-холестеринемию),атеросклероз, ожирение и другие нарушения, связанные с липидным метаболизмом, и их осложнения в отношении энергетического гомеостаза. Вышеуказанные способы включают введение пациенту, нуждающемуся в лечении, фармацевтически эффективного количества композиции, включающей соединение по изобретению. Примеры состояний, которые можно лечить с помощью агониста дельта-PPAR, включают, без ограничения, диабет, сердечно-сосудистые заболевания, метаболический синдром X, гиперхолестеринемию, гипо-HDL-холестеринемию, гипер-LDL-холестеринемию, дислипидемию, атеросклероз и ожирение. Дислипидемия включает гипертриглицеридемию и смешанную гиперлипидемию. Например, дислипидемия (включая гиперлипидемию) может представлять собой одно или более из следующих состояний: с низким уровнем HDL (35 или 40 мг/дл), с высоким уровнем триглицеридов (200 мг/дл) и с высоким уровнем LDL (150 мг/дл). Дополнительные воплощения и преимущества согласно данному изобретению будут видны из нижеприводимых подробного обсуждения, примеров и формулы изобретения. Подробное описание данного изобретения Данное изобретение относится к фармацевтическим композициям, содержащим соединения формулы (I), как указано выше в разделе Краткое изложение сущности изобретения, и способам их применения. Предпочтительные соединения согласно данному изобретению являются агонистами дельта-PPAR,имеющими по меньшей мере одну и предпочтительно две или три из следующих характеристик при введении пациентам с гиперхолестеринемией, гипертриглицеридемией, с низким уровнем HDL-C, ожирением, диабетом и/или метаболическим синдромом X: 1) увеличение уровня HDL-C; 2) уменьшение количества триглицеридов; 3) уменьшение количества свободных жирных кислот и 4) снижение уровней инсулина. Повышение уровней HDL-C и снижение уровней триглицеридов является благоприятным в случае сердечно-сосудистого заболевания. Кроме того, снижение уровня триглицеридов и свободных жирных кислот способствует уменьшению ожирения и ослабляет или предотвращает диабет. В соответствии с одним аспектом согласно данному изобретению соединения согласно данному изобретению являются двойными агонистами PPAR; иначе говоря, они являются и агонистами дельтаPPAR, и агонистами альфа-PPAR, предпочтительно где эффективность соединений ЕС 50 относительно дельта-PPAR составляет менее чем 0,2 мкМ и эффективность относительно альфа-PPAR составляет менее чем 3 мкМ. Например, более предпочтительными двойными агонистами альфа-дельта-PPAR являются такие соединения, которые имеют эффективность ЕС 50 относительно дельта-PPAR, составляющую менее чем 0,03 мкМ, и где эффективность относительно альфа-PPAR составляет менее чем 1 мкМ. В соответствии с другим аспектом согласно данному изобретению соединения согласно данному изобретению являются общими PPAR-агонистами, а именно соединениями, имеющими PPAR-альфа-,PPAR-дельта- и PPAR-гамма-агонистическую активность, предпочтительно где эффективность ЕС 50 относительно дельта-PPAR составляет менее чем 0,2 мкМ; эффективность ЕС 50 относительно альфа-PPAR составляет менее чем 3 мкМ и эффективность ЕС 50 относительно гамма-PPAR составляет менее чем 1 мкМ. Более предпочтительные общие PPAR-агонисты имеют эффективность ЕС 50 в случае дельтаPPAR, которая составляет менее чем 0,03 мкМ; эффективность ЕС 50 в случае альфа-PPAR составляет менее чем 1 мкМ и эффективность ЕС 50 в случае гамма-PPAR составляет менее чем 0,7 мкМ. Дельта-PPAR, экспрессируемый повсеместно, может действовать как рецептор мембранного канала, регулирующий экспрессию/активность других ядерных рецепторов, таких как другие PPAR. Например, показано, что дельта-PPAR блокирует опосредованный PPAR адипогенез и экспрессию ацил-СоАоксидазы; также показано, что он связан с сорепрессорами ядерных рецепторов SMRT (латентный медиатор рецепторов ретиноидов и тиреоидного гормона), SHARP (SMART и ассоциированный с гистондезацетилазой белок-репрессор) и HDAC (гистон-дезацетилаза). Таким образом, состояния, прямо опосредованные этими ядерными рецепторами, такие как ожирение и диабет типа II, могут быть косвенно-3 013403 опосредованы дельта-PPAR (см., например, Shi и др., 2002, Proc. Natl. Acad. Sci., США, 99(5): 2613-2618). Некоторые аспекты согласно данному изобретению относятся к лечению гипертриглицеридемии,повышению уровней HDL, снижению уровней LDL и/или снижению общего холестерина. Предпочтительно способы лечения связаны с улучшениями в отношении меры, продолжительности или степени побочных эффектов, таких как отек, обычно ассоциированный с другими существующими терапевтическими методами. Далее, данное изобретение раскрывается ниже. Описание изобретения систематизировано следующим образом: А) термины; В) соединения; С) синтез; D) готовая лекарственная форма и ее введение; Е) применение; F) биологические примеры; G) другие воплощения и формула изобретения. А. Термины Термин пациент, как используется в данном описании, относится к животному, предпочтительно к млекопитающему, наиболее предпочтительно к человеку, который является объектом для лечения, наблюдения или эксперимента. Термин фармацевтически эффективное количество, как используется в данном описании, означает такое количество активного соединения или фармацевтического агента, которое вызывает биологический или медицинский ответ в тканевой системе животного или человека, определяемый исследователем, ветеринаром, врачом или другим клиницистом, который включает ослабление, предотвращение,лечение или замедление появления или развития симптомов излечиваемого заболевания или нарушения. Состояния, прямо или косвенно опосредованные PPAR, включают, но не ограничиваясь этим, диабет, нефропатию, невропатию, ретинопатию, синдром поликистоза яичника, гипертензию, ишемию,удар, синдром раздраженной толстой кишки, воспаление, катаракту, сердечно-сосудистые заболевания,метаболический синдром X, гипер-LDL-холестеринемию, дислипидемию (включая гипертриглицеридемию, гиперхолестеринемию, смешанную гиперлипидемию и гипо-HDL-холестеринемию), атеросклероз,ожирение и другие заболевания, связанные с липидным метаболизмом, и их осложнения в отношении энергетического гомеостаза. За исключением иначе указанного, как используется в данном описании, и используется ли индивидуально или как часть замещающей группы, термины алкил и алкокси включают линейные или разветвленные цепи,имеющие 1-9 атомов углерода, такие как C1-6, C1-4, C3-8, С 2-5 или любой другой диапазон, и, за исключением иначе указанного, включают как замещенные, так и незамещенные остатки. Например, C1-6-алкил включает метил,этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 3-(2-метил)бутил, 2-пентил,2-метилбутил, неопентил, н-гексил, 2-гексил и 2-метилпентил. Алкокси-радикалы образуются из вышеописанных алкильных групп с линейной или разветвленной цепью. Термины алкил и алкокси включают незамещенные или замещенные остатки с одним или более замещениями, такие как с 1-5, 1-3 или 2-4 заместителями. Заместители могут быть одинаковыми (дигидрокси, диметил), подобными (хлор, фтор) или различными (хлорбензил- или аминометилзамещенные). Примеры замещенных алкилов включают галогеналкил (такой как фторметил, хлорметил, дифторметил, перхлорметил, 2-бромэтил, трифторметил и 3-иодциклопентил), гидроксиалкил(такой как гидроксиметил, гидроксиэтил, 2-гидроксипропил), аминоалкил (такой как аминометил, 2-аминоэтил,3-аминопропил и 2-аминопропил), алкоксилалкил, нитроалкил, алкилалкил, цианоалкил, фенилалкил, феноксиалкил, тиоалкилалкил (такой как MeS-алкил), тиофенилалкил (такой как PhS-алкил), карбоксиалкил и т.д. Ди(С 1-3-алкил)аминогруппа включает независимо выбираемые алкильные группы для образования, например метилпропиламино и изопропилметиламино, причем, кроме того, диалкиламиногруппы имеют две одинаковые алкильные группы, такие как диметиламино или диэтиламино. Термин алкенил включает углеводородные радикалы с линейной или разветвленной цепью, как описано выше, по меньшей мере с одной двойной углерод-углеродной связью (sp2). Алкенилы включают этенил (или винил), проп-1-енил, проп-2-енил (или аллил), изопропенил (или 1-метилвинил), бут-1-енил,бут-2-енил, бутадиенилы, пентенилы, гекса-2,4-диенил и т.д. Углеводородные радикалы, включающие смесь двойных связей и тройных связей, такие как 2-пентен-4-инил, в данном описании классифицируют как алкинилы. Алкенил включают циклоалкенил. Цис- и транс- или (Е)-и (Z)-формы включены в рамки данного изобретения. Алкенил может быть замещен одним или более заместителями, включая, но не ограничиваясь этим, цианоалкенил и тиоалкенил. Термин алкинил включает углеводородные радикалы с линейной или разветвленной цепью, как описано выше, по меньшей мере с одной тройной углерод-углеродной связью (sp). Алкинилы включают этинил, пропинилы, бутинилы и пентинилы. Углеводородные радикалы, включающие смесь двойных связей и тройных связей, такие как 2-пентен-4-инил, в данном описании классифицируют как алкинилы. Термин алкинил не включает циклоалкинил. Термин Ас, как используется в данном описании, и используется ли индивидуально или как часть замещающей группы, означает ацетил (CH3CO-). Термин ацил, как используется в данном описании,относится к заместителю, который включает карбонильную группу (C=O) и одну или более алкильных или алкиленовых групп. Например, С 2-4-ацил включает, без ограничения, ацетил, СН 3 СН 2-(C=O)-СН 2- и СН 3 СН 2 СН 2(C=O)-. Термин галоген или гало включает иод, бром, хлор и фтор.-4 013403 Подразумевают, что значение любого заместителя или радикала в конкретном положении в молекуле не зависит от его значений в другом положении в этой молекуле. Подразумевают, что заместители и замещенные структуры в соединениях согласно данному изобретению могут быть выбраны любым специалистом в данной области для получения соединений, которые являются химически стабильными и которые можно без труда синтезировать способами, известными в уровне техники, а также способами,изложенными в данном описании. Когда химические фрагменты являются комбинированными, такими как этоксиметил или фенилэтил, термин описывается в направлении от периферии к месту присоединения к остальной части молекулы. Например, этоксиметил представляет собой CH3CH2OCH2- и фенилэтил представляет собой фенильную группу, связанную через -СН 2 СН 2- с остальной частью молекулы (а не фенильную группу, связанную с молекулой с помощью группы CH3CH2 в качестве заместителя фенила). Там, где используют скобки, они указывают периферическое замещение. Подразумевают, как используется в данном описании, что термин композиция означает продукт,включающий определенные ингредиенты в определенных количествах, а также любой продукт, который получают, прямо или косвенно, из комбинаций определенных ингредиентов в определенных количествах. Соединения согласно данному изобретению далее описываются в следующем разделе. В. Соединения Настоящее изобретение относится к соединениям формулы (I), как описано выше. Примеры соединений формулы (I) включают такие соединения, где (а) X означает S или О; (b) X означает ковалентную связь; (с) X означает О; (d) Y означает О; (е) Y означает S; (f) Z означает О; (g) Z означает СН или СН 2;C1-3-алкокси, замещенного галогеном; (l) R1 и R2 независимо выбирают из Н, C1-3-алкила, C1-3-алкокси, F,Cl и Br; (m) R1 и R2 независимо выбирают из Н, метила, метокси, F и Cl; (n) R4 выбирают из С 1-4-алкила,замещенного галогеном, и C1-3-алкокси, замещенного галогеном; (о) R3 независимо выбирают из Н, F иCl; (p) R4 независимо выбирают из трифторметила, фторметила, дифторметила, хлордифторметила, дихлорфторметила, фторметокси, дифторметокси, хлордифторметокси, дихлорфторметокси и трифторметокси; (q) R3 выбирают из Н, Cl, Br и I и R4 выбирают из Н, Cl и метила; (r) R7 выбирают из C1-7-алкила,C1-6-алкокси, С 2-7-алкенила, С 2-7-алкенилокси, С 2-7-алкинила, С 2-7-алкинилокси, C3-7-циклоалкила,C3-7-циклоалкокси, C1-6-алкокси-C1-6-алкила, С 1-5-алкокси-С 1-5-алкокси и C3-7-циклоалкилокси-С 1-7-алкокси;-5 013403 Конкретные примеры соединений формулы (I) включают Соединение 1. 2-Метил-2-2-метил-4-[2-(4-трифторметилфеноксиметил)бутилсульфанил]феноксипропионовая кислота. Примеры предпочтительных соединений включают таковые, описанные в нижеприведенной табл. 1. Таблица 1 Фармацевтические композиции согласно данному изобретению включают фармацевтические композиции, содержащие фармацевтически приемлемый носитель и соединение формулы (I). Когда соединения согласно данному изобретению имеют по меньшей мере один хиральный центр,они могут, соответственно, существовать в виде энантиомеров. Когда соединения имеют два или более хиральных центра, они могут дополнительно существовать в виде диастереомеров. Подразумевают, что все такие изомеры и их смеси входят в рамки объема настоящего изобретения. Кроме того, некоторые из кристаллических форм соединений могут существовать в виде полиморфов и подразумевают, что они-7 013403 входят в рамки объема настоящего изобретения. Кроме того, некоторые из соединений могут образовывать сольваты с водой (т.е. гидраты) или обычными органическими растворителями, и подразумевают,что такие сольваты также входят в рамки объема данного изобретения. Данное изобретение относится к фармацевтически приемлемым солям описанных соединений. Фармацевтически приемлемые соли включают карбоксилатные соли, аддитивные соли аминокислот, которые находятся в рамках приемлемого соотношения польза/риск, являются фармакологически эффективными и пригодными для контакта с тканями пациентов без чрезмерной токсичности, раздражения или аллергического ответа. Типичные соли включают гидробромид, гидрохлорид, сульфат, бисульфат, нитрат, ацетат, оксалат, валерат, олеат, пальмитат, стеарат, лаурат, борат, бензоат, лактат, фосфат,тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, гликогептонат, лактиобионат и лаурилсульфонат. Они могут включать катионы щелочных и щелочно-земельных металлов, таких как натрий, калий, кальций и магний, а также нетоксический аммоний, четвертичный аммоний и аминокатионы, как, например, тетраметиламмоний, метиламин, триметиламин и этиламин. См., например, статью S.M. Berge и др., "Pharmaceutical Salts", J. Pharm. Sci., 1977, 66: 1-19, которая включена в данное описание путем ссылки. С. Синтез Данное изобретение относится к способам получения описанных соединений в соответствии со стандартными способами органического синтеза, а также к матриксным или комбинаторным способам синтеза. На схемах A-G представлены направления синтеза. Используя эти схемы, указанные ниже, и примеры, любой специалист в данной области может разработать аналогичные или подобные способы для получения соединений, которые входят в рамки данного изобретения. Эти способы являются типичными из предпочтительных схем синтеза, но не могут быть истолкованы как ограничивающие объем данного изобретения. Квалифицированному специалисту в данной области понятно, что синтез соединений согласно настоящему изобретению можно осуществлять путем приобретения промежуточных или защищенных промежуточных продуктов, описанных в случае любой из схем, представленных согласно данному описанию. Квалифицированному специалисту в данной области далее понятно, что во время любого из процессов получения соединений согласно настоящему изобретению может быть необходимо и/или желательно защищать чувствительные или реакционноспособные группы, относящиеся к любой из молекул. Этого можно достигать с помощью стандартных защитных групп, таких как описанные в руководстве"Protective Groups in Organic Synthesis", John WileySons, 1991. Эти защитные группы можно удалять на подходящей стадии, используя известные из уровня техники способы. Когда способы получения соединений согласно данному изобретению приводят к образованию смеси стереоизомеров, эти изомеры можно разделять путем стандартных методов, таких как препаративная хроматография. Соединения могут быть получены в рацемической форме, или отдельные энантиомеры можно получать или путем энантиоспецифического синтеза, или путем разделения. Соединения,например, могут быть разделены на составляющие их энантиомеры путем стандартных способов, таких как образование диастереомерных пар за счет образования соли. Соединения также могут быть разделены путем образования диастереомерных сложных эфиров или амидов, затем путем хроматографического разделения и удаления хиральной вспомогательной группы. Альтернативно, соединения можно разделять, используя ВЭЖХ-колонку с хиральной фазой. Примеры описанных путей синтеза включают примеры 1-7. Соединения, аналогичные целевым соединениям этих примеров, можно получать согласно подобным путям синтеза. Описанные соединения пригодны для фундаментальных исследований и в качестве фармацевтических агентов, как описывается в следующем разделе. Общее руководство Предпочтительный синтез соединений формулы (I) показан на схемах 1-9. Аббревиатуры или акронимы, используемые в данном описании, включают: АсОН (ледяная уксусная кислота); DCC (1,3-дициклогексилкарбодиимид); DCE (1,2-дихлорэтан); DIC (2-диметиламиноизопропилхлоридгидрохлорид); DIEA (диизопропилэтиламин); DMAP (4-(диметиламино)пиридин); ДМФА (диметилформамид); EDC (1-(3-диметиламинопропил)-3-этилкарбодиимид); EtOAc (этилацетат);-8 013403 Схема 1 Синтез промежуточного продукта 1-D В соответствии со схемой 1, где R1, R2, R5 и R6 имеют значения, как описано выше, фенол 1-А из множества коммерчески доступных (как, например, 3-метилфенол, 2-этилфенол, 2-пропилфенол, 2,3 диметилфенол, 2-хлорфенол, 2,3-дихлорфенол, 2-бромфенол и 2-аминофенол) алкилируют до образования этилового эфира феноксиуксусной кислоты 1-В с помощью подходящего эфира галогенуксусной кислоты, такого как этиловый эфир бромуксусной кислоты или этиловый эфир 2-бром-2-метилпропионовой кислоты, в присутствии соответствующего основания, такого как Cs2CO3, K2CO3 или NaH, в подходящем растворителе, таком как CH3CN или ТГФ. Сульфирование этилового эфира феноксиуксусной кислоты 1-В с помощью соответствующего сульфирующего агента, такого как хлорсульфоновая кислота, происходит селективно в пара-положении с образованием этилового эфира 4-хлорсульфонилфеноксиуксусной кислоты 1-С. Превращения сульфонилхлорида 1-С в бензолтиол 1-D достигают, используя металл в качестве восстановителя, такой как олово или цинк, в кислой среде, такой как этанол или диоксан. Схема 2 Синтез соединения Ia На схеме 2 R7-замещенный диэтилмалонат 2-А восстанавливают до пропан-1,3-диола 2-В, используя подходящий восстановитель, такой как литийалюминийгидрид или диизобутилалюминийгидрид. Путем реакции Мицунобу соединения 2-В с фенолом 2-С получают соединение 2-D, используя триарилфосфин, такой как трифенилфосфин, и азодикарбонильный реагент, такой как диизопропилазодикарбоксилат, в подходящем растворителе, таком как ТГФ. Этиловый эфир феноксиуксусной кислоты 2-Е получают в две стадии: (1) превращение спирта 2-D в мезилат в стандартных условиях, используя метансульфонилхлорид и триэтиламин, в соответствующем растворителе, таком как CH2Cl2; и (2) алкилирование бензолтиола 1-D, полученного согласно вышеприведенной схеме 1, с помощью промежуточного мезилата, используя подходящее основание, такое как Cs2CO3, K2CO3 или NaH, в соответствующем растворителе, таком как CH3CN или ТГФ, в атмосфере азота. В стандартных условиях омыления этиловый эфир феноксиуксусной кислоты 2-Е превращают в кислоту Ia в атмосфере азота. Предпочтительные условия гидролиза включают использование NaOH в качестве основания в водно-спиртовой системе в качестве растворителя, такой как вода-метанол, или использование LiOH в качестве основания в более мягкой системе вода-ТГФ. Согласно схеме 3 энантиомерно чистую фенилуксусную кислоту 3-А из множества коммерчески доступных (как, например, (S)-(+)-2-фенилпропионовая кислота, (R)-(-)-2-фенилпропионовая кислота,(S)-(+)-2-фенилмасляная кислота, (R)-(-)-2-фенилмасляная кислота, (+)-3-метил-2-фенилмасляная кислота, (S)-(+)-2-фенилянтарная кислота и (R)-(-)-2-фенилянтарная кислота) восстанавливают до спирта, используя боран, и спирт затем защищают в виде ацетата 3-В в стандартных условиях, известных в данной области. Окисление фенильной группы в соединении 3-В до кислоты 3-С осуществляют, используя каталитическое количество хлорида рутения и большого избытка периодата натрия в системе смешанного растворителя, такой как CH3CN-CCl4-H2O. Кислоту 3-С превращают в спирт 3-Е в четыре стадии: (1) метилирование кислоты 3-С, используя (триметилсилил)диазометан в качестве метилирующего агента; (2) и (3) замена ацетатной гидроксилзащитной группы в соединении 3-С на трет-бутилдиметилсилилоксигруппу в соединении 3-Е в стандартных условиях, хорошо известных в данной области; и (4) восстановление сложного метилового эфира, используя соответствующий восстановитель, такой как диизобутилалюминийгидрид. Этиловый эфир феноксиуксусной кислоты 3-F получают в две стадии: (1) превращение спирта 3-Е в мезилат в стандартных условиях, используя метансульфонилхлорид и триэтиламин, в соответствующем растворителе, таком как CH2Cl2; и (2) алкилирование бензолтиола 1-D, полученного согласно вышеприведенной схеме 1, с помощью промежуточного мезилата, используя подходящее основание, такое какCs2CO3, K2CO3 или NaH, в соответствующем растворителе, таком как CH3CN или ТГФ, в атмосфере азота. После освобождения гидроксильной группы путем удаления трет-бутилдиметилсилилоксигруппы в соединении 3-F спирт 3-G превращают в соединение 3-Н путем введения во взаимодействие с фенолом 2-С в условиях реакции Мицунобу. Предпочтительные условия включают использование триарилфосфина, такого как трифенилфосфин, и азодикарбонильного реагента, такого как диизопропилазодикарбоксилат, в подходящем растворителе, таком как ТГФ. В стандартных условиях омыления этиловый эфир феноксиуксусной кислоты 3-Н превращают в кислоту Ia1 в атмосфере азота. Предпочтительные условия гидролиза включают использование NaOH в качестве основания в водно-спиртовой системе в качестве растворителя, такой как вода-метанол, или использование LiOH в качестве основания в более мягкой системе вода-ТГФ. На схеме 4 бензолтиол 1-D димеризуют до фенилдисульфида 4-А в присутствии соответствующего окислителя, такого как манганат бария. Путем реакции по Мицунобу 2-гидроксиметилпропан-1,3-диола 4-В с фенолом 2-С получают соединение 4-С, используя триарилфосфин, такой как трифенилфосфин, и азодикарбонильный реагент,такой как диизопропилазодикарбоксилат, в подходящем растворителе, таком как ТГФ. Образование связи углерод-сера в соединении 4-D осуществляют путем реакции по Мицунобу диола 4-С с фенилдисульфидом 4-А, используя три-н-бутилфосфин и пиридин. Путем третьей реакции по Мицунобу соединения 4-D с ацетонцианогидрином спирт 4-D превращают в цианосоединение 4-Е в стандартных условиях реакции Мицунобу. Как обычно, щелочной гидролиз этилового эфира феноксиуксусной кислоты 4-Е приводит к получению кислоты Ia2. Схема 5 Синтез соединения Ia3 Как показано на схеме 5, где R означает алкил или арил, простой алкиловый эфир соединения 5-А можно получать путем алкилирования спирта 4-D, промежуточного продукта, получаемого согласно вышеприведенной схеме 4, с помощью множества алкилирующих агентов, таких как алкилтрифторметансульфонаты или алкилгалогениды, в присутствии подходящего основания, такого как гидрид натрия или бис(триметилсилил)амид натрия. Подобным образом, ариловый эфир можно синтезировать по реакции Мицунобу из соединения 4-D при использовании множества различных коммерчески доступных замещенных фенолов. Наконец, омыление сложного этилового эфира соединения 5-А в стандартных условиях приводит к получению кислоты Ia3. Схема 6 Синтез соединения Ia4 В соответствии со схемой 6 путем реакции по Мицунобу (R)-(+)-глицидола или (S)-(-)-глицидола- 11013403 или рацемического глицидола 6-А с фенолом 2-С получают эпоксид 6-В, используя триарилфосфин, такой как трифенилфосфин, и азодикарбонильный реагент, такой как диизопропилазодикарбоксилат, в подходящем растворителе, таком как ТГФ. Раскрытие эпоксидного цикла соединения 6-В с помощью бензолтиола 1-D в присутствии каталитического количества тетрабутиламмонийфторида приводит к получению спирта 6-С. Простой алкиловый эфир 6-D можно получать путем алкилирования спирта 6-С с помощью множества алкилирующих агентов, таких как алкилтрифторметансульфонаты или алкилгалогениды, в присутствии подходящего основания, такого как гидрид натрия или бис(триметилсилил)амид натрия, в подходящем растворителе, таком как ТГФ или ДМФА. Подобным образом, ариловый эфир 6-D можно синтезировать путем реакции по Мицунобу соединения 6-С с множеством различных коммерчески доступных замещенных фенолов, используя трифенилфосфин и соответствующий азодикарбонильный реагент, такой как 1,1'-(азодикарбонил)дипиперидин или диэтилазодикарбоксилат. Наконец, омыление этилового эфира 6-D в стандартных условиях приводит к получению кислоты Ia4. Схема 7 Синтез промежуточного продукта 7-Е В соответствии со схемой 7 (4-гидроксифенил)уксусную кислоту 7-А из множества коммерчески доступных (как, например, 3-бром-4-гидроксифенилуксусная кислота, 3-хлор-4-гидроксифенилуксусная кислота, 3-фтор-4-гидроксифенилуксусная кислота, 4-гидрокси-3-метоксифенилуксусная кислота и 4-гидрокси-3-нитрофенилуксусная кислота) метилируют с получением метилового эфира (4-гидроксифенил)уксусной кислоты 7-В в метаноле, в присутствии каталитического количества подходящей кислоты, такой как серная кислота или соляная кислота. Фенол 7-В превращают в метиловый эфир (4-диметилтиокарбамоилоксифенил)уксусной кислоты 7-С путем взаимодействия с диметилтиокарбамоилхлоридом в присутствии некоторых соответствующих оснований, таких как триэтиламин и 4-(диметиламино)пиридин. При высокой температуре в предпочтительном диапазоне 250-300 С соединение 7-С перегруппируют в метиловый эфир (4-диметилкарбамоилсульфанилфенил)уксусной кислоты 7-D в растворителе с высокой температурой кипения, таком как тетрадекан. Путем обработки подходящим основанием, таким как метоксид натрия, соединение 7-D превращают в метиловый эфир (4-меркаптофенил)уксусной кислоты 7-Е. Схема 8 Синтез соединения Ib1 В соответствии со схемой 8, где R означает алкил, эпоксид 8-В получают путем обработки фенола 2-С соответствующим основанием, таким как карбонат цезия, а затем путем алкилирования 2-хлорметилоксираном 8-А. Раскрытие эпоксидного цикла в соединении 8-В с помощью бензолтиола 7-Е, получаемого согласно вышеприведенной схеме 7, в присутствии каталитического количества тетрабутиламмонийфторида, приводит к получению спирта 8-С. Простой алкиловый эфир 8-D можно получать путем алкилирования спирта 8-С множеством алкилирующих агентов, таких как алкилтрифторметансульфонаты или алкилгалогениды, в присутствии подходящего основания, такого как гидрид натрия или- 12013403 бис(триметилсилил)амид натрия, в подходящем растворителе, таком как ТГФ или ДМФА. Наконец,омыление сложного метилового эфира 8-D в стандартных условиях приводит к получению кислоты Ib1. Схема 9 Синтез соединения Ic1 На схеме 9, где R имеет значение, как указано выше, альдегид 9-В можно получать в две стадии путем метилирования кислоты 9-А, используя (триметилсилил)диазометан в качестве метилирующего агента, а затем восстановления промежуточного сложного метилового эфира с помощью соответствующего восстановителя, такого как диизобутилалюминийгидрид. Альдегид 9-В превращают в эпоксид 9-С путем введения во взаимодействие с диметилсульфонийметилидом, который получают in-situ обработкой триметилсульфонийиодида сильным основанием, таким как ДМСО-анион. Раскрытие эпоксидного цикла в соединении 9-С с помощью бензолтиола 1-D в присутствии каталитического количества тетрабутиламмонийфторида приводит к получению спирта 9-D. Простой алкиловый эфир 9-Е можно получать путем алкилирования спирта 9-D множеством алкилирующих агентов, таких как алкилтрифторметансульфонаты или алкилгалогениды, в присутствии подходящего основания, такого как гидрид натрия или бис(триметилсилил)амид натрия, в подходящем растворителе, таком как ТГФ или ДМФА. Наконец,омыление сложного этилового эфира 9-Е в стандартных условиях приводит к получению кислоты Ic1. Примеры Пример А. 2-Метил-4-[2-(4-трифторметилфеноксиметил)бутилсульфанил]феноксиуксусная кислота. В соответствии со схемой А 1, в колбу, содержащую хлорсульфоновую кислоту (15,0 мл,226 ммоль), при температуре 4 С медленно добавляют этил(2-метилфенокси)ацетат A1a (10,0 г,51,6 ммоль). Смесь перемешивают при температуре 4 С в течение 30 мин и при комнатной температуре в течение 2 ч и затем выливают в смесь воды со льдом. Осажденное белое твердое вещество отфильтровывают, промывают водой и сушат в вакууме в течение ночи, получая 14,0 г (93%) соединения A1b в виде белого твердого вещества. 1H ЯМР (300 МГц, CDCl3)7,87-7,84 (м, 2 Н), 6,80 (д, J=9,5 Гц, 1 Н), 4,76 (с, 2 Н), 4,29 (кв, J=7,1 Гц,2 Н), 2,37 (с, 3H), 1,31 (т, J=7,1 Гц, 3H); МС (ионизация электронным распылением, ES) m/z: 315 (M+Na+). К раствору соединения A1b (4,70 г, 16,1 ммоль) в EtOH (20 мл) добавляют 4,0 М раствор HCl в диоксане (20 мл), затем порциями добавляют порошкообразное олово с размером частиц 100 меш (9,80 г,- 13013403 82,6 ммоль). Смесь кипятят с обратным холодильником в течение 2 ч, выливают в смесь CH2Cl2 со льдом(100 мл) и отфильтровывают. Фильтрат разделяют и водный слой экстрагируют CH2Cl2. Объединенные органические фазы промывают водой, сушат и концентрируют, получая 3,56 г (98%) соединения A1c в виде желтого масла. 1 К суспензии литийалюминийгидрида (101 мг, 2,66 ммоль) в ТГФ (3 мл) при температуре 0 С по каплям добавляют диэтилэтилмалонат А 2 а (250 мг, 1,33 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 ч, гасят водой (0,1 мл) и 5 н. раствором NaOH (0,2 мл), разбавляют водой (0,6 мл), отфильтровывают через целит и промывают твердое вещество смесью МеОН/CH2Cl2. Фильтрат сушат, концентрируют и очищают с помощью колоночной хроматографии, получая 110 мгH ЯМР (300 МГц, CDCl3)3,79 (дд, J=10,7, 3,9 Гц, 2 Н), 3,64 (дд, J=10,7, 7,5 Гц, 2 Н), 3,27 (с, 2 Н),1,67 (м, 1 Н), 1,29 (м, 2 Н), 0,94 (т, J=7,5 Гц, 3H); МС (ES), m/z: 127 (M+Na+). К смеси соединения А 2b (108 мг, 1,04 ммоль), трифторметилфенола (130 мг, 0,802 ммоль) и трифенилфосфина (210 мг, 0,802 ммоль) в ТГФ (3 мл) при температуре 0 С добавляют диизопропилазодикарбоксилат (162 мг, 0,802 ммоль). Смесь перемешивают при комнатной температуре в течение ночи, разбавляют водой и экстрагируют Et2O (3 раза). Экстракты сушат, концентрируют и подвергают колоночной хроматографии, получая 134 мг (67%) соединения А 2 с. 1Et3N (0,162 мл, 1,16 ммоль) и метансульфонилхлорид (93 мг, 0,81 ммоль). Смесь перемешивают при температуре 0 С в течение 30 мин и при комнатной температуре в течение 1 ч и разбавляют насыщенным раствором NaHCO3. Органический слой отделяют и водный слой экстрагируют CH2Cl2 (3 раза). Объединенные органические фазы сушат и концентрируют, получая мезилат. Смесь вышеуказанного мезилата, этилового эфира (4-меркапто-2-метилфенокси)уксусной кислотыA1c (197 мг, 0,872 ммоль) и Cs2CO3 (472 мг, 1,45 ммоль) в CH3CN (5 мл) перемешивают при комнатной температуре в течение 3 ч. Добавляют воду и смесь экстрагируют Et2O. Объединенные органические слои сушат, концентрируют и подвергают колоночной хроматографии (EtOAc/гексан: 1/10), получая 213 мг (81% за две стадии) соединения A2d. 1H ЯМР (300 МГц, CDCl3)7,50 (д, J=8,6 Гц, 2 Н), 7,19 (д, J=1,8 Гц, 1 Н), 7,15 (дд, J=8,4, 2,2 Гц, 1 Н),6,89 (д, J=8,6 Гц, 2 Н), 6,56 (д, J=8,4 Гц, 1 Н), 4,56 (с, 2 Н), 4,25 (кв, J=7,1 Гц, 2 Н), 4,01 (м, 2 Н), 3,00 (д,J=6,4 Гц, 2 Н), 2,21 (с, 3H), 1,96 (м, 1 Н), 1,59 (м, 2 Н), 1,28 (т, J=7,1 Гц, 3H), 0,94 (т, J=7,5 Гц, 3H); МС (ES),m/z: 479 (M+Na+). Общая методика 2. Гидролиз сложных этиловых и метиловых эфиров. К раствору соединения A2d (134 мг, 0,294 ммоль) в ТГФ (2 мл) в атмосфере азота добавляют 1,0 М раствор LiOH (0,58 мл, 0,58 ммоль). Смесь перемешивают в течение 2 ч, подкисляют 1 М раствором HCl и экстрагируют EtOAc (3 раза). Экстракты сушат, концентрируют и очищают с помощью колоночной хроматографии (CH2Cl2/МеОН: 10/1), получая 113 мг (90%) 2-метил-4-[2-(4-трифторметилфеноксиметил)бутилсульфанил]феноксиуксусной кислоты. 1Cs2CO3 (15,7 г, 48,2 ммоль) в диоксане (8 мл) кипятят с обратным холодильником в течение 3-4 ч и затем оставляют охлаждаться до комнатной температуры. Добавляют воду и Et2O, органическую фазу отделяют и водную фазу экстрагируют Et2O. Объединенные органические слои сушат, концентрируют и подвергают колоночной хроматографии (CH2Cl2/гексан: 1/1), получая 8,40 г (80%) соединения В 1. 1(0,44 мл, 0,44 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 1,5 ч, нагревают при температуре 60 С в течение 1 ч, концентрируют и очищают с помощью колоночной хроматографии, получая 4,45 г (85%) соединения В 2. 1(дд, J=13,7, 5,1 Гц, 1 Н), 3,04 (дд, J=13,9, 6,5 Гц, 1 Н), 2,92 (д, J=4,2 Гц, 1 Н), 2,23 (с, 3H), 1,28 (т, J=7,1 Гц,3H); МС (ES) m/z: 467 (M+Na+). Общая методика 3. Алкилирование спиртов. К суспензии NaH (20 мг, 0,50 ммоль, 60%-ная в минеральном масле) в ТГФ (1 мл) добавляют раствор соединения В 2 (222 мг, 0,500 ммоль) в ТГФ (1 мл) при комнатной температуре. Спустя 30 мин вносят C2H5I (234 мг, 1,50 ммоль). Реакционную смесь перемешивают в течение ночи, разбавляют водой и экстрагируют Et2O. Экстракты сушат, концентрируют и очищают с помощью колоночной хроматографии (EtOAc/гексан: 1/6), получая соединение B3. 1H ЯМР (300 МГц, CDCl3)7,51 (д, J=8,6 Гц, 2 Н), 7,24 (д, J=1,7 Гц, 1 Н), 7,19 (дд, J=8,4, 2,2 Гц, 1 Н),6,91 (д, J=8,6 Гц, 2 Н), 6,57 (д, J=8,4 Гц, 1 Н), 4,57 (с, 2 Н), 4,25 (кв, J=7,1 Гц, 2 Н), 4,15 (дд, J=9,9, 4,3 Гц,1 Н), 4,07 (дд, J=9,9, 5,1 Гц, 1 Н), 3,76 (м, 1 Н), 3,61 (кв, J=7,0 Гц, 2 Н), 3,13-3,11 (м, 2 Н), 2,23 (с, 3H), 1,29 (т,J=7,1 Гц, 3H), 1,18 (т, J=7,0 Гц, 3H); МС (ES) m/z: 495 (M+Na+). Следуя общей методике 2 согласно примеру А получают 4-[2-этокси-3-(4-трифторметилфенокси)пропилсульфанил]-2-метилфеноксиуксусную кислоту (выход 92%). 1 К раствору соединения A2d (245 мг, 0,54 ммоль) в ТГФ (3 мл) при температуре -78 С по каплям добавляют 1 н. раствор литийбис(триметилсилил)амида в ТГФ (0,54 мл, 0,54 ммоль). Спустя 30 мин добавляют метилтрифторметансульфонат (0,061 мл, 0,54 ммоль). Реакционную смесь оставляют постепенно нагреваться до температуры 0 С в течение 1 ч, гасят насыщенным водным раствором NaHCO3 и экстрагируют Et2O (3 раза). Экстракты сушат, концентрируют и подвергают колоночной хроматографии, получая 64,5 мг (25%) соединения С 1. 1H ЯМР (300 МГц, CDCl3)7,51 (д, J=8,8 Гц, 2 Н), 7,18 (д, J=1,8 Гц, 1 Н), 7,12 (дд, J=8,5, 1,8 Гц, 1 Н),6,90 (д, J=8,8 Гц, 2 Н), 6,55 (д, J=8,5 Гц, 1 Н), 4,65 (кв, J=6,8 Гц, 1 Н), 4,19 (кв, J=7,1 Гц, 2 Н), 4,01 (м, 2 Н),2,99 (д, J=6,5 Гц, 2 Н), 2,20 (с, 3H), 1,96 (м, 1 Н), 1,60 (д, J=6,8 Гц, 3H), 1,58 (м, 2 Н), 1,24 (т, J=6,1 Гц, 3H),0,94 (т, J=7,5 Гц, 3H); МС (ES) m/z: 493 (M+Na+). Соединение 1 (выход 91%) получают, следуя общей методике 2 согласно примеру А. 1 К раствору этилового эфира 2-бром-2-метилпропионовой кислоты (8,27 мл, 64 ммоль) и о-метилфенола (7,60 г, 70,2 ммоль) в диоксане (100 мл) добавляют Cs2CO3 (31,25 г, 96 ммоль). Смесь кипятят с обратным холодильником при температуре 100 С в течение 4 ч. После охлаждения растворитель выпаривают в вакууме. Остаток растворяют в диэтиловом эфире и затем промывают 1 н. раствором NaOH.- 16013403 После сушки раствор концентрируют, получая 9,69 г (68%) соединения D1. 1ClSO3H (15,2 мл, 0,229 моль) медленно добавляют к соединению D1 (11,3 г, 0,051 моль) при температуре 0 С. Смесь оставляют нагреваться до комнатной температуры и перемешивают в течение 1 ч. После перемешивания реакционную смесь выливают на лед. Твердое вещество отфильтровывают и сушат в вакууме, получая 7,7 г (47%) соединения D2. 1(4 М раствор, 7,8 мл, 31,2 ммоль) и порошкообразное олово (3,7 г, 31,2 ммоль). Смесь кипятят с обратным холодильником в течение 3 ч и затем выливают на лед. Водный раствор экстрагируют CH2Cl2(3 раза по 50 мл). Органические слои объединяют и сушат над Na2SO4. После фильтрации раствор концентрируют, получая 3,37 г (100%) соединения D3. 1DIAD (634 мг, 3,14 ммоль) в CH2Cl2 (10 мл) добавляют Ph3P (833 мг, 3,14 ммоль) в атмосфере азота. После перемешивания в течение ночи раствор разбавляют диэтиловым эфиром (40 мл) и промывают 1 н. раствором NaOH. Органический слой затем сушат и концентрируют, получая 107 мг сырого продукта. К вышеуказанному промежуточному продукту (530 мг, 2,54 ммоль) в CH2Cl2 (10 мл) при температуре 0 С добавляют Et3N (0,75 мл, 5,4 ммоль) и затем MeSO2Cl (0,32 мл, 4,15 ммоль). Смесь перемешивают при температуре 0 С в течение 10 мин и затем при комнатной температуре в течение 2 ч. После концентрирования сырой продукт очищают с помощью колоночной хроматографии (50% CH2Cl2 в гексанах), получая 188 мг сырого промежуточного продукта. Вышеуказанный сырой промежуточный продукт (183 мг, 0,56 ммоль), соединение D3 (171 мг, 0,67 ммоль) и Cs2CO3 (438 мг, 1,34 ммоль) смешивают в CH3CN (10 мл). После перемешивания в течение 1,5 ч к раствору добавляют смесь вода-диэтиловый эфир, водную часть экстрагируют диэтиловым эфиром (3 раза по 20 мл). Органические экстракты объединяют и сушат над Na2SO4. После фильтрации фильтрат концентрируют и затем очищают с помощью колоночной хроматографии (EtOAc/гексаны: 1/20), получая 108 мг (10% за 3 стадии) соединения D4. 1 Соединение 3 2-4-[2-Этокси-3-(4-трифторметилфенокси)пропилсульфанил]-2-метилфенокси-2 метилпропионовая кислота Схема Е(348 мг, 1,37 ммоль) в безводном ТГФ (10 мл) добавляют TBAF (137 мг, 0,137 ммоль). После перемешивания в течение ночи раствор концентрируют и продукт очищают с помощью колоночной хроматографии (20% смесь EtOAc в гексанах), получая 191 мг (31%) соединения Е 1. 1 Этиловый эфир 2-4-[2-этокси-3-(4-трифторметилфенокси)пропилсуль фанил]-2-метилфенокси-2-метилпропионовой кислоты К раствору соединения Е 1 (148 мг, 0,313 ммоль) в безводном ТГФ (1 мл) при температуре -78 С добавляют NaHMDS (1 М раствор в ТГФ, 0,313 мл, 0,313 ммоль) и EtOTs (55,8 мг, 0,313 ммоль). После перемешивания в течение 10 мин раствор затем оставляют нагреваться до температуры 0 С и продолжают перемешивать при температуре 0 С в течение одного часа. Добавляют диэтиловый эфир и раствор промывают водой и затем насыщенным солевым раствором. После сушки над Na2SO4 и концентрирования сырой продукт очищают с помощью колоночной хроматографии (EtOAc/гексаны: 20/1), получая 80 мг 2-4-[2-Этокси-3-(4-трифторметилфенокси)пропилсульфанил]-2-метил фенокси-2-метилпропионовая кислота К раствору соединения Е 2 (65 мг, 0,13 ммоль) в ТГФ (1 мл) добавляют NaOH (480 мг, 12 ммоль) в смеси МеОН-H2O (2:1 по объему, 3 мл). После перемешивания в течение 2 ч раствор подкисляют 1 н. раствором HCl. Раствор затем экстрагируют диэтиловым эфиром (3 раза по 10 мл). Органические слои объединяют и сушат над Na2SO4 и концентрируют. Сырой продукт затем очищают с помощью колоночной хроматографии (10% МеОН в CH2Cl2), получая 40 мг (65%) соединения 3. 1D. Готовая лекарственная форма и введение. Соединения согласно настоящему изобретению являются агонистами дельта-PPAR и, следовательно, пригодны для лечения или ингибирования развития опосредованных дельта-PPAR состояний, таких как диабет, сердечно-сосудистые заболевания, метаболический синдром X, гиперхолестеринемия, гипоHDL-холестеринемия, гипер-LDL-холестеринемия, дислипидемия, атеросклероз, ожирение и их осложнений. Например, диабетические осложнения включают такие состояния, как невропатия, нефропатия и ретинопатия. Данное изобретение относится к способу лечения субъекта с опосредованным дельта-PPAR заболеванием, причем вышеуказанный способ включает введение субъекту терапевтически эффективного количества фармацевтической композиции, содержащей соединение согласно данному изобретению. Данное изобретение также относится к способу лечения или ингибирования развития диабета или снижения толерантности к глюкозе у субъекта, причем способ включает введение субъекту терапевтически эффективного количества фармацевтической композиции, содержащей соединение согласно данному изобретению. Соединения согласно настоящему изобретению могут быть использованы для получения различных фармацевтических форм для целей введения. Для получения этих фармацевтических композиций эффективное количество отдельного соединения в форме аддитивной соли кислоты или основания, в качестве активного ингредиента, тщательно перемешивают с фармацевтически приемлемым носителем. Носитель можно выбирать из широкого разнообразия форм, в зависимости от формы лекарственного средства, желательного для введения. Эти фармацевтические композиции желательны в унифицированной лекарственной форме, пригодной предпочтительно для перорального введения или парентеральной инъекции. Например, при получении композиций в пероральной лекарственной форме можно использовать любую из обычных фармацевтических сред. Они включают воду, гликоли, масла, спирты и т.п. в случае пероральных жидких композиций, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие вещества,дезинтегрирующие агенты и т.п., в случае порошков, пилюль, капсул и таблеток. Принимая во внимание легкость их введения, таблетки и капсулы представляют собой наиболее предпочтительную унифицированную лекарственную форму, в случае которой, как правило, используют твердые фармацевтические носители. Для парентеральных композиций носитель обычно включает стерильную воду, по меньшей мере, большей частью, несмотря на то что могут быть включены другие ингредиенты, например, для способствования растворимости. Можно получать, например, растворы для инъекций, в которых носитель включает насыщенный солевой раствор, раствор глюкозы или смесь насыщенного солевого раствора и раствора глюкозы. Также можно получать суспензии для инъекций, в случае которых можно использовать соответствующие жидкие носители, суспендирующие агенты и т.п. В композициях, пригодных для чрескожного введения, носитель необязательно включает повышающий пенетрацию агент и/или пригодный смачиватель, необязательно комбинированный с пригодными добавками любой природы в незначительных количествах, причем эти добавки не оказывают значительного вредного воздействия на кожу. Такие добавки могут облегчать введение в кожу и/или могут быть полезны для получения желательных композиций. Эти композиции можно вводить различными путями, например в виде трансдермального пластыря, в виде притирания, в виде мази. Аддитивные соли кислот соединений формулы (I) вследствие их повышенной растворимости в воде за счет соответствующей основной формы более пригодны для получения водных композиций. Особенно преимущественным является получение вышеуказанных фармацевтических композиций в унифицированной лекарственной форме для легкости введения и единообразия дозировки. Унифицированная лекарственная форма, как используется в данном описании, относится к физически дискретным единицам, пригодным в качестве унифицированных доз, каждая единица которых содержит заранее определенное количество активного ингредиента, рассчитанное на получение желательного терапевтического эффекта, в сочетании с необходимым фармацевтическим носителем. Примерами таких унифицированных лекарственных форм являются таблетки (включая таблетки с насечкой или таблетки с нанесенным покрытием), капсулы, пилюли, пакетированные порошки, облатки, растворы или суспензии для инъекций, дозы лекарственных средств в объеме полной чайной ложки, дозы лекарственных средств в объеме полной столовой ложки и т.п., и их разделенные на множество частей формы. Фармацевтически приемлемые аддитивные соли кислот включают терапевтически активные нетоксичные аддитивные соли кислот описанных соединений. Последние можно без труда получать путем обработки основной формы соответствующей кислотой. Соответствующие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, как, например, соляная кислота или бромводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные кислоты; или органические кислоты, такие как, например, уксусная кислота, пропионовая кислота, гидроксиуксусная кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, этансульфоно- 19013403 вая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, цикламиновая кислота, салициловая кислота, п-аминосалициловая кислота, пальмовая кислота и подобные кислоты. Термин аддитивная соль также относится к сольватам описанных соединений, а также к образованным ими солям. Такие сольваты представляют собой, например, гидраты, алкоголяты и т.п. Наоборот, солевая форма может быть превращена в форму свободного основания путем обработки щелочью. Стереоизомерные формы включают все возможные изомерные формы, которые могут иметь соединения формулы (I). За исключением иначе указанного или определенного, химическое название соединений относится к смеси всех возможных стереохимически изомерных форм, причем вышеуказанные смеси содержат все возможные диастереомеры и энантиомеры основной молекулярной структуры. Еще,в частности, стереогенные центры могут иметь (R)- или (S)-конфигурацию; заместители двухвалентных циклических насыщенных радикалов могут иметь или цис- или транс-конфигурацию. Данное изобретение включает стереохимически изомерные формы, включая диастереоизомеры, а также их смеси в любой пропорции в случае описанных соединений. Описанные соединения также могут существовать в таутомерных формах. Подразумевают, что такие формы, хотя и подробно не указанные выше и в последующих формулировках, входят в рамки объема настоящего изобретения. Квалифицированный специалист по лечению нарушений или состояний, опосредованных дельтаPPAR, может без труда определять эффективное суточное количество из результатов теста, представленного в дальнейшем, или из другой информации. Вообще, предполагают, что терапевтически эффективная доза должна составлять от 0,001 до 5 мг/кг массы тела, более предпочтительно от 0,01 до 0,5 мг/кг массы тела. Может быть пригодным введение терапевтически эффективной дозы в виде двух, трех, четырех или более субдоз в соответствующие интервалы в течение суток. Вышеуказанные субдозы можно получать в виде унифицированных лекарственных форм, например, содержащих от 0,05 до 250 мг или 750 мг, и, в частности, от 0,5 до 50 мг активного ингредиента на унифицированную лекарственную форму. Примеры включают 2, 4, 7, 10, 15, 25 и 35 мг лекарственной формы. Соединения согласно данному изобретению также можно использовать для получения пролонгированно высвобождающихся или подкожных или в виде трансдермальных пластырей готовых лекарственных форм. Раскрытое соединение также можно использовать для получения лекарственной формы в виде спрея или других локальных или вводимых путем ингаляции готовых лекарственных средств. Точная дозировка и частота введения зависят от используемого конкретного соединения формулы(I), конкретного излечиваемого состояния, тяжести излечиваемого состояния, возраста, массы тела и общего физического состояния конкретного пациента, а также другого вида лечения, которому пациент может подвергаться, как хорошо известно квалифицированному специалисту в данной области. Более того, очевидно, что вышеуказанное эффективное суточное количество можно снижать или увеличивать в зависимости от ответной реакции излечиваемого пациента и/или в зависимости от оценки врача, назначающего препараты с соединениями согласно настоящему изобретению. Диапазоны эффективного суточного количества, указанные в данном описании, следовательно, являются только ориентировочными. Следующий раздел включает подробную информацию по применению описанных соединений и композиций. Е. Применение. Соединения согласно настоящему изобретению являются фармацевтически активными, например, в качестве агонистов дельта-PPAR и предпочтительно в качестве двойных агонистов альфа/дельта-PPAR. В соответствии с одним аспектом данного изобретения соединения являются предпочтительно селективными агонистами дельта-PPAR, имеющими индекс активности (например, эффективность дельта-PPAR выше, чем эффективность альфа/гамма-PPAR) равным 10 или более и предпочтительно 15, 25, 30, 50 или 100 или более. В соответствии с другим аспектом соединения являются двойными агонистами альфаPPAR и дельта-PPAR. Согласно данному изобретению описанные соединения и композиции пригодны для ослабления симптомов, связанных с лечением и предотвращением следующих состояний и заболеваний: гиперлипидемия фазы I, доклиническая гиперлипидемия, гиперлипидемия фазы II, гипертензия, CAD (коронарное артериальное заболевание), атеросклероз, коронарная болезнь сердца, сердечно-сосудистое заболевание,гиперхолестеремия и гипертриглицеридемия, диабет типа II, резистентность к инсулину, сниженная толерантность к глюкозе, дислипидемия и низкий уровень HDL-C. Предпочтительные соединения согласно данному изобретению пригодны для снижения уровней в сыворотке липопротеинов низкой плотности(LDL), липопротеинов средней плотности (IDL) и/или LDL низкой плотности и других атерогенных молекул или молекул, являющихся причиной атеросклеротических осложнений, уменьшая, таким образом,сердечно-сосудистые осложнения. Предпочтительные соединения также пригодны для повышения уровней в сыворотке липопротеинов высокой плотности (HDL), снижения уровней в сыворотке триглицеридов, LDL и/или свободных жирных кислот. Также желательно снижать содержание глюкозы в плазме в состоянии натощак (FPG)/HbA1c. Опосредованные альфа-PPAR заболевания включают синдром X (или метаболический синдром),дислипидемию, высокое кровяное давление, ожирение, резистентность к инсулину, пониженное содержание глюкозы натощак, диабет типа II, атеросклероз, неалкогольный стеатогепатит, гиперхолестерине- 20013403 мию, гипертриглицеридемию и низкий уровень HDL-C. В соответствии с одним аспектом данного изобретения описанные соединения можно использовать в случае способа лечения или ингибирования развития состояния, опосредованного дельта-PPAR, и, необязательно, дополнительного состояния, опосредованного альфа-PPAR, причем вышеуказанный способ включает введение пациенту, нуждающемуся в лечении, фармацевтически эффективного количества композиции согласно данному изобретению. Другой аспект согласно данному изобретению представляет собой способ применения, где состояние, опосредованное дельта-PPAR, выбирают из группы, состоящей из гиперлипидемии, атеросклероза,сердечно-сосудистого заболевания, гиперхолестеремии, диабета типа II, резистентности к инсулину и пониженной толерантности к глюкозе, и других состояний, указанных в данном описании; и состояние,опосредованное альфа-PPAR, выбирают из синдрома X (или метаболического синдрома), дислипидемии,высокого кровяного давления, ожирения и пониженного содержания глюкозы натощак, резистентности к инсулину, диабета типа II и других состояний, указанных в данном описании. Дальнейший аспект согласно данному изобретению представляет собой способ лечения по меньшей мере одного опосредованного дельта-PPAR состояния и по меньшей мере одного опосредованного альфа-PPAR состояния у пациента, причем вышеуказанный способ включает введение пациенту, нуждающемуся в лечении, фармацевтически эффективного количества композиции согласно данному изобретению. Данное изобретение также относится к фармацевтическим композициям, которые включают, без ограничения, одно или более описанных соединений и фармацевтически приемлемый носитель или эксципиент. 1. Дозировки. Квалифицированный специалист в данной области может определить, в соответствии с известными способами, соответствующую дозировку для пациента при рассмотрении факторов, таких как возраст,масса тела, общее состояние здоровья, тип подвергаемых лечению симптомов и наличие других видов лечения. Вообще, эффективное количество может составлять от 0,1 до 1000 мг/кг в сутки, предпочтительно от 1 до 300 мг/кг массы тела, и суточные дозы могут находиться в диапазоне от 10 до 5000 мг для взрослого субъекта с нормальной массой тела. Капсулы, таблетки или другие готовые лекарственные формы (такие как жидкие или покрытые пленкой таблетки) могут быть массой от 5 до 200 мг, как, например, 10, 15, 25, 35, 50, 60 и 100 мг, и могут быть введены в соответствии с указанными способами. 2. Готовые лекарственные формы. Унифицированные лекарственные формы включают таблетки, капсулы, пилюли, порошки, гранулы, водные и неводные пероральные растворы и суспензии, и парентеральные растворы, упакованные в контейнеры, подходящие для подразделения на отдельные дозы. Унифицированные лекарственные формы также могут быть использованы для различных способов введения, включая готовые лекарственные формы с контролируемым высвобождением, такие как подкожные имплантаты. Способы введения включают пероральный, ректальный, парентеральный (внутривенный, внутримышечный, подкожный), интрацистернальный, интравагинальный, внутрибрюшинный, интравезикальный, локальный (капли, порошки,мази, гели или кремы), и путем ингаляции (спрей для ротовой полости или для назального применения). Парентеральные готовые лекарственные формы включают фармацевтически приемлемые водные или неводные растворы, дисперсии, суспензии, эмульсии и стерильные порошки для их получения. Примеры носителей включают воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль), растительные масла и инъецируемые органические сложные эфиры, такие как этилолеат. Жидкое состояние можно сохранять путем использования покрытия, такого как лецитин, поверхностно-активное вещество или путем поддерживания соответствующего размера частиц. Носители для твердых лекарственных форм включают (а) наполнители или порофоры; (b) связующие вещества; (с) увлажнители; (d) дезинтегрирующие агенты, (е) замедлители растворения; (f) ускорители абсорбции; (g) адсорбенты; (h) смазывающие вещества; (i) буферные агенты и (j) пропелленты. Композиции также могут содержать вспомогательные средства, такие как консерванты, смачиватели, эмульгаторы и дозирующие средства; противомикробные агенты, такие как парабены, хлорбутанол,фенол и сорбиновая кислота; изотонические агенты, такие как сахар или хлорид натрия; пролонгирующие абсорбцию агенты, такие как моностеарат алюминия и желатин; и увеличивающие абсорбцию агенты.F. Биологические примеры. Трансфекционный анализ PPAR-рецепторов. Клетки HEK293 культивировали в модифицированной по способу Дульбекко среде Игла(DMEM/F12), дополненной 10% фетальной телячьей сыворотки (FBS) и глутамином (Invitrogen), и инкубировали в инкубаторе в атмосфере с 5% CO2 при температуре 37 С. Клетки подвергали котрансфекции,используя реагент DMRIE-С (Invitrogen) в свободной от сыворотки среде (Opti-MEM, Invitrogen) с помощью двух экспрессируемых млекопитающими плазмид, причем одна содержит ДНК-последовательность, кодирующую лигандсвязывающие домены или PPAR, - или -, слитые с доменом, связывающим ДНК GAL4 дрожжей, а другая содержит промоторную последовательность GAL4 дрожжей (UAS), сшитую с репортером кДНК люциферазы светляка. На следующий день среду заменяли средой DMEM/F12, дополненной- 21013403 5% обработанной активированным углем сыворотки (Hyclone) и глутамином. Спустя 6 ч клетки обрабатывали трипсином и высевали в 96-луночные планшеты при плотности 50000 клеток на лунку и инкубировали в течение ночи, как указано выше. Затем клетки обрабатывали тестируемыми соединениями или средой и инкубировали в течение 18-24 ч, как указано выше. Активность репортера люциферазы измеряли, используя набор для анализа Steady-Glo Luciferase Assay Kit, выпускаемый фирмой Promega. РеагентDMRIE-C приобретали у фирмы GIBCO, регистрационный номер по каталогу 10459-014. Среду со сниженным содержанием сыворотки OPTI-MEM I приобретали у фирмы GIBCO (регистрационный номер по каталогу 31985). Набор для анализа Steady-Glo Luciferase Assay Kit приобретали у фирмы Promega (регистрационный номер по каталогу Е 254 В). Ряд типичных соединений получен и протестирован с получением результатов в области in vitro. Ниже представлены соединения и данные; в некоторых случаях, где указано несколько значений ЕС 50,выполнено несколько измерений. Естественно, различные соединения формулы (I) могут иметь или не иметь активности, идентичные активности любого соединения, указанного ниже. Таблица 2 Данные in vitroG. Другие воплощения. Признаки и принципы данного изобретения проиллюстрированы в обсуждении, примерах и формуле изобретения в данном описании. Различные адаптации и модификации данного изобретения должны быть очевидны квалифицированному специалисту в данной области и такие другие воплощения также входят в рамки данного изобретения. Публикации, цитированные в данном описании, включены во всей их полноте путем ссылки. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) где X выбирают из ковалентной связи, S или О;R1 и R2 независимо выбирают из Н, C1-3-алкила, C1-3-алкокси и галогена;R4 независимо выбирают из Н, галогена, циано, гидрокси, ацетила, C1-5-алкила, C1-5-алкила, замещенного галогеном, С 1-4-алкокси и C1-4-алкокси, замещенного галогеном, при условии, что R3 и R4 оба не означают Н;R5 и R6 независимо выбирают из Н, C1-8-алкила и замещенного C1-8-алкила, при условии, что R5 и R6 оба не означают Н;R8 означает Н, когда представляет собой группу, выбираемую из -СН=, -СН 2-,представляет собой груп-СН 2-СН 2-, -СН 2-СН= и -СН=СН-; или R8 отсутствует, когда пу, выбираемую из =СН-, =СН-СН 2- и =СН-СН=; иn означает 1 или 2; или его фармацевтически приемлемая соль. 2. Соединение по п.1, где X означает S или О. 3. Соединение по п.1, где X означает ковалентную связь. 4. Соединение по п.2, где X означает О. 5. Соединение по п.1, где Y означает О. 6. Соединение по п.1, где Y означает S.- 22013403 7. Соединение по п.1, где Z означает О. 8. Соединение по п.1, где Z означает СН или СН 2. 9. Соединение по п.1, где представляет собой группу -СН 2- или -СН 2-СН 2-. 10. Соединение по п.9, где представляет собой группу -СН 2-. 11. Соединение по п.1, где представляет собой группу =СН-, -СН=, =СН-СН 2-,-СН 2-СН=, =СН-СН= или -СН=СН-. 12. Соединение по п.1, где R4 выбирают из галогена, C1-4-алкила, C1-4-алкила, замещенного галогеном, C1-3-алкокси и C1-3-алкокси, замещенного галогеном. 13. Соединение по п.1, где R1 и R2 независимо выбирают из Н, C1-3-алкила, C1-3-алкокси, F, Cl и Br. 14. Соединение по п.13, где R1 и R2 независимо выбирают из Н, метила, метокси, F и Cl. 15. Соединение по п.1, где R4 выбирают из C1-4-алкила, замещенного галогеном, и C1-3-алкокси, замещенного галогеном. 16. Соединение по п.12, где R3 независимо выбирают из Н, F и Cl. 17. Соединение по п.12, где R4 независимо выбирают из трифторметила, фторметила, дифторметила, хлордифторметила, дихлорфторметила, фторметокси, дифторметокси, хлордифторметокси, дихлорфторметокси и трифторметокси. 18. Соединение по п.1, где R3 выбирают из Н, Cl, Br и I и R4 выбирают из Н, Cl и метила. 19. Соединение по п.1, где R7 выбирают из C1-7-алкила, C1-6-алкокси, С 2-7-алкенила, С 2-7-алкенилокси,С 2-7-алкинила, С 2-7-алкинилокси, C3-7-циклоалкила, C3-7-циклоалкокси, C1-6-алкокси-C1-6-алкила, С 1-5-алкоксиС 1-5-алкокси и C3-7-циклоалкилокси-С 1-7-алкокси. 20. Соединение по п.1, где R7 выбирают из С 3-7-циклоалкил-C1-7-алкила, C3-7-циклоалкил-С 1-7-алкокси и C3-7-циклоалкилокси-С 1-6-алкила. 21. Соединение по п.1, где R8 означает Н. 22. Соединение по п.1, где R3 выбирают из Н, F и Cl и R4 выбирают из F, Cl, метила, фторметила,дифторметила, фторметокси, дифторметокси, трифторметила, трифторметокси и метокси. 23. Соединение по п.1, где R1 выбирают из Н, метила, Cl и метокси и R2 выбирают из Н, Cl и метила. 24. Соединение по п.23, где X означает ковалентную связь. 25. Соединение по п.23, где X означает ковалентную связь, Y означает S и Z означает О. 26. Соединение по п.1, где X означает О и Y означает О. 27. Соединение по п.1, где X означает О и Y означает S. 28. Соединение по п.1, где Y означает О и Z означает О. 29. Соединение по п.1, где Y означает S и Z означает О. 30. Соединение по п.1, где R8 означает Н и R7 выбирают из C1-7-алкила, C1-6-алкокси, С 2-7-алкенила,С 2-7-алкенилокси, C1-6-алкокси-С 1-6-алкила и С 1-5-алкокси-С 1-5-алкокси. 31. Соединение по п.30, где R7 выбирают из C1-5-алкила, C1-4-алкокси, C2-5-алкенила, C2-5-алкенилокси и С 1-5-алкокси-С 1-5-алкокси. 32. Соединение по п.30, где R7 выбирают из C1-3-алкила, C1-3-алкокси, C2-4-алкенила, С 2-4-алкенилокси и C1-3-алкокси-C1-3-алкокси. 33. Соединение по п.30, где R7 выбирают из метокси, этокси, пропокси, изопропокси, пропенилокси, изопропенилокси, этоксиметокси, метоксиметокси, метоксиметила, метоксиэтила, этоксиметила и этоксиэтила. 34. Соединение по п.1, где R5 и R6 независимо означают C1-4-алкил. 35. Соединение по п.34, где R5 и R6 означают метил. 36. Соединение по п.1, гдеR4 выбирают из F, Cl, метила, CF3, OCF3 и метокси. 47. Соединение по п.44, где n=1. 48. Соединение по п.45, где R5 означает C1-3-алкил и R8 означает Н. 49. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически приемлемый носитель. 50. Способ лечения или ингибирования развития опосредованного дельта-PPAR состояния, выбираемого из группы, состоящей из диабета, нефропатии, невропатии, ретинопатии, синдрома поликистоза яичника, гипертензии, ишемии, удара, синдрома раздраженной толстой кишки, воспаления, катаракты,сердечно-сосудистых заболеваний, метаболического синдрома X, гипер-LDL-холестеринемии, гипертриглицеридемии, гиперхолестеринемии, смешанной гиперлипидемии, гипо-HDL-холестеринемии, атеросклероза, ожирения и их осложнений, включающий введение пациенту, нуждающемуся в лечении,фармацевтически эффективного количества композиции по п.49. или его фармацевтически приемлемая соль. 52. Соединение, выбранное из группы, состоящей из или его фармацевтически приемлемая соль. или его фармацевтически приемлемая соль. 54. Фармацевтическая композиция, содержащая соединение по пп.51, 52 или 53 и фармацевтически приемлемый носитель. 55. Способ лечения или ингибирования развития опосредованного дельта-PPAR состояния, выбираемого из группы, состоящей из диабета, нефропатии, невропатии, ретинопатии, синдрома поликистоза яичника, гипертензии, ишемии, удара, синдрома раздраженной толстой кишки, воспаления, катаракты,сердечно-сосудистых заболеваний, метаболического синдрома X, гипер-LDL-холестеринемии, гипертриглицеридемии, гиперхолестеринемии, смешанной гиперлипидемии, гипо-HDL-холестеринемии, атеросклероза, ожирения и их осложнений, включающий введение пациенту, нуждающемуся в лечении,фармацевтически эффективного количества композиции по п.54.
МПК / Метки
МПК: C07C 59/68, A61K 31/185, C07C 323/10, C07C 323/20, C07C 323/62
Метки: 4-((феноксиалкил)тио)феноксиуксусные, аналоги, кислоты
Код ссылки
<a href="https://eas.patents.su/27-13403-4-fenoksialkiltiofenoksiuksusnye-kisloty-i-analogi.html" rel="bookmark" title="База патентов Евразийского Союза">4-((феноксиалкил)тио)феноксиуксусные кислоты и аналоги</a>
4-((феноксиалкил)тио)феноксиуксусные кислоты и их аналоги

Номер патента: 12241
Опубликовано: 28.08.2009
Авторы: Чжан Жуй, Ко Джи-Хун, Ван Айхуа, Диэнджелис Алан Р.
МПК: A61K 31/192, A61K 31/381, A61K 31/277...
Метки: аналоги, кислоты, 4-((феноксиалкил)тио)феноксиуксусные
Формула / Реферат:
1. Соединение формулы (I) где X выбран из ковалентной связи или О, Y представляет собой S или О; ---W--- представляет собой группу, выбранную из -CH=, -СН2-; Z выбран из О, CH и СН2, при условии, что когда Y представляет собой О, Z представляет собой О; R1 представляет собой H или C1-3алкил; R2 представляет собой H или галоген; R3 представляет собой H; R4 представляет собой С1-5алкил, замещенный галогеном; R5 представляет собой необязательно...
4-((феноксиалкил)тио)феноксиуксусные кислоты и аналоги

Номер патента: 12849
Опубликовано: 30.12.2009
Авторы: Чжан Жуй, Ван Айхуа, Ко Джи-Хун, Демарест Кит Т., Диэнджелис Алан, Пелтон Патрисия
МПК: C07C 323/20, C07C 323/10, A61K 31/185...
Метки: кислоты, 4-((феноксиалкил)тио)феноксиуксусные, аналоги
Формула / Реферат:
1. Соединение формулы (I) где X выбирают из ковалентной связи, S и O; Y означает S или О; Z выбирают из О или CH2 при условии, что, когда Y означает О, Z означает О; R1 и R2 независимо выбирают из H, C1-3-алкила, C1-3-алкокси и галогена; R3 означает H, галоген, гидрокси, С1-5-алкил или С1-4-алкокси; R4 означает H, галоген, циано, С1-5-алкил, С1-5-алкил, замещенный галогеном, гидрокси, С2-4-ацил, С1-4-алкокси или С1-4-алкокси, замещенный...
4- ((феноксиалкил)тио) феноксиуксусные кислоты и аналоги

Номер патента: 9119
Опубликовано: 26.10.2007
Авторы: Ванг Айхуа, Чжан Жуй, Диэнджелис Алан Р., Куо Джи-Хонг
МПК: A61K 31/192, A61K 31/196, A61K 31/277...
Метки: феноксиуксусные, феноксиалкил)тио, кислоты, аналоги
Формула / Реферат:
1. Соединение формулы (I) где X выбирают из ковалетной связи, S и О; Y означает S или О; Z означает О или CH2, при условии, что когда Y означает О, тогда Z означает О; R1 и R2 независимо выбирают из Н, C1-3-алкила, C1-3-алкоксила, галогена и NRaRb, где Ra и Rb независимо означают Н или C1-3-алкил; R3 и R4 независимо выбирают из Н, галогена, цианогруппы, C1-5-алкила, гидроксила, С2-4-ацила, C1-4-алкоксила и NRcRd, где Rc и Rd независимо означают...
Жирные кислоты со средней длиной цепи, глицериды и аналоги как факторы, способствующие выживанию и активации нейтрофилов

Номер патента: 7322
Опубликовано: 25.08.2006
Авторы: Бараб Жан, Ганьон Лин, Лорэн Пьер, Пенни Кристофер, Зашари Було
МПК: A61K 31/20, A61K 31/18, A61K 31/194...
Метки: глицериды, цепи, длиной, факторы, выживанию, жирные, средней, активации, кислоты, способствующие, нейтрофилов, аналоги
Формула / Реферат:
1. Применение соединения для производства лекарственного средства для лечения состояний, выбранных из миелосупрессии, ранений, трансплантации костного мозга и нейропении, причем указанное соединение имеет любую из формул I, II, IIа, III и IIIа в которых R1 означает С7-11 алкил; А и В означают независимо водород или R2 означает Н или C1-4 алкил, М означает одновалентный катион металла (n=1) или дикатион (n=2); Y означает О или NH и Z означает...
Жирные кислоты со средней длиной цепи, глицериды и аналоги в качестве стимуляторов эритропоэза

Номер патента: 9939
Опубликовано: 28.04.2008
Авторы: Зашари Було, Ганьон Лин, Пенни Кристофер, Лорэн Пьер
МПК: A61K 31/20
Метки: качестве, жирные, аналоги, средней, стимуляторов, длиной, кислоты, глицериды, эритропоэза, цепи
Формула / Реферат:
1. Применение композиции, содержащей соединение любой из формул I, II и IIa или их комбинацию где каждый R1 независимо представляет собой С7-11алкил; А и В независимо представляют собой H или CO-R1; R2 представляет собой H или С1-4алкил; M представляет собой монокатион металла (k=1) или дикатион (k=2); Y представляет собой O или NH; для производства медикамента для стимуляции эритропоэза. 2. Применение по п.1, где композиция содержит по меньшей...