Способ производства органических карбонатов
















Формула / Реферат
1. Способ получения диарилкарбоната, включающий:
(a) осуществление взаимодействия реагентов в нескольких реакционных зонах, включая первичную и вторичную реакционные зоны;
(b) подвод к указанной первичной реакционной зоне диалкилкарбоната и ароматического гидроксисоединения;
(c) осуществление взаимодействия реагентов в первичной реакционной зоне в двухфазной системе и реакционных условиях, способствующих образованию алкиларилкарбоната;
(d) переэтерификацию данного диалкилкарбоната с ароматическим гидроксисоединением в первичной реакционной зоне в присутствии твердого катализатора, выбранного из группы, включающей от двух до четырех элементов групп IV, V и VI Периодической таблицы, нанесенных на пористый материал, который имеет поверхностные гидроксильные группы;
(e) извлечение потока двухфазного продукта из первичной реакционной зоны;
(f) отделение потока двухфазного продукта со стадии (е) для извлечения парообразного алкилового спирта и жидкого алкиларилкарбоната;
(g) диспропорционирование алкиларилкарбоната во вторичной реакционной зоне в двухфазной системе в присутствии твердого катализатора, выбранного из группы, включающей от двух до четырех элементов групп IV, V и VI Периодической таблицы, нанесенных на пористый материал, который имеет поверхностные гидроксильные группы;
(h) извлечение потока двухфазного продукта из вторичной реакционной зоны и
(i) отделение потока двухфазного продукта со стадии (h) для извлечения парообразного компонента, включающего алкиларилкарбонат, и жидкого продукта, включающего диарилкарбонат.
2. Способ по п.1, в котором диалкилкарбонат представляет собой диэтилкарбонат (ДЭК), ароматическое гидроксисоединение представляет собой фенол, алкиларилкарбонат представляет собой этилфенилкарбонат (ЭФК) и диарилкарбонат представляет собой дифенилкарбонат (ДФК).
3. Способ по п.2, в котором на стадии (g) мольное отношение фенола к ДФК находится в диапазоне от 0,05 до 10.
4. Способ по п.1, в котором твердый катализатор на стадии (d) представляет собой оксиды, гидроксиды, оксигидроксиды, алкоксиды или их смеси элементов из групп IV, V и VI.
5. Способ по п.4, в котором носитель катализатора включает обработанный кремнезем.
6. Способ по п.4, в котором твердый катализатор на стадии (g) представляет собой оксиды, гидроксиды, оксигидроксиды, алкоксиды или их смеси элементов из групп IV, V и VI.
7. Способ по п.5, в котором твердый катализатор на стадии (d) выбирают из группы, состоящей из металлических алкоксидов и смешанных металлических алкоксидов групп IV и V.
8. Способ по п.4, в котором твердый катализатор на стадии (d) представляет собой TiO2/Nb2O5, нанесенный на обработанный кремнезем, причем указанный обработанный кремнезем имеет менее чем 0,05 мас.% Na.
9. Способ по п.1, в котором твердый катализатор на стадии (g) представляет собой оксиды, гидроксиды, оксигидроксиды, алкоксиды или их смеси элементов из групп IV, V и VI.
10. Способ по п.9, в котором носитель катализатора включает обработанный кремнезем.
11. Способ по п.10, в котором твердый катализатор на стадии (g) выбирают из группы, состоящей из металлических алкоксидов и смешанных металлических алкоксидов групп IV и V.
12. Способ по п.9, в котором твердый катализатор на стадии (g) представляет собой TiO2/Nb2O5, нанесенный на обработанный кремнезем, причем указанный обработанный кремнезем имеет менее чем 0,05 мас.% Na.
13. Способ по п.1, в котором процесс на стадии (g) выполняют в присутствии ароматического гидроксисоединения при мольном отношении ароматического гидроксисоединения к ДФК в диапазоне от 0,05:1 до 10:1.
14. Способ по п.13, в котором процесс на стадии (g) выполняют в присутствии воды в количестве до 0,3 мас.%.
15. Способ по п.1, в котором процесс на стадии (d) выполняют в присутствии ароматического гидроксисоединения при мольном отношении ароматического гидроксисоединения к ДЭК выше 0,2.
16. Композиция твердого катализатора диспропорционирования или переэтерификации, выбранного из группы, состоящей из оксидов, гидроксидов, оксигидроксидов и алкоксидов от двух до четырех элементов из групп IV, V и VI Периодической таблицы, нанесенного на пористый материал, который имеет поверхностные гидроксильные группы.
17. Композиция твердого катализатора по п.16, в которой указанный носитель предварительно подвергают обработке, чтобы увеличить число содержащихся на нем гидроксильных групп.
18. Композиция твердого катализатора по п.17, в которой указанная обработка включает контактирование указанного носителя с раствором основания.
19. Композиция твердого катализатора по п.18, в которой указанный носитель включает кремнезем.
Текст
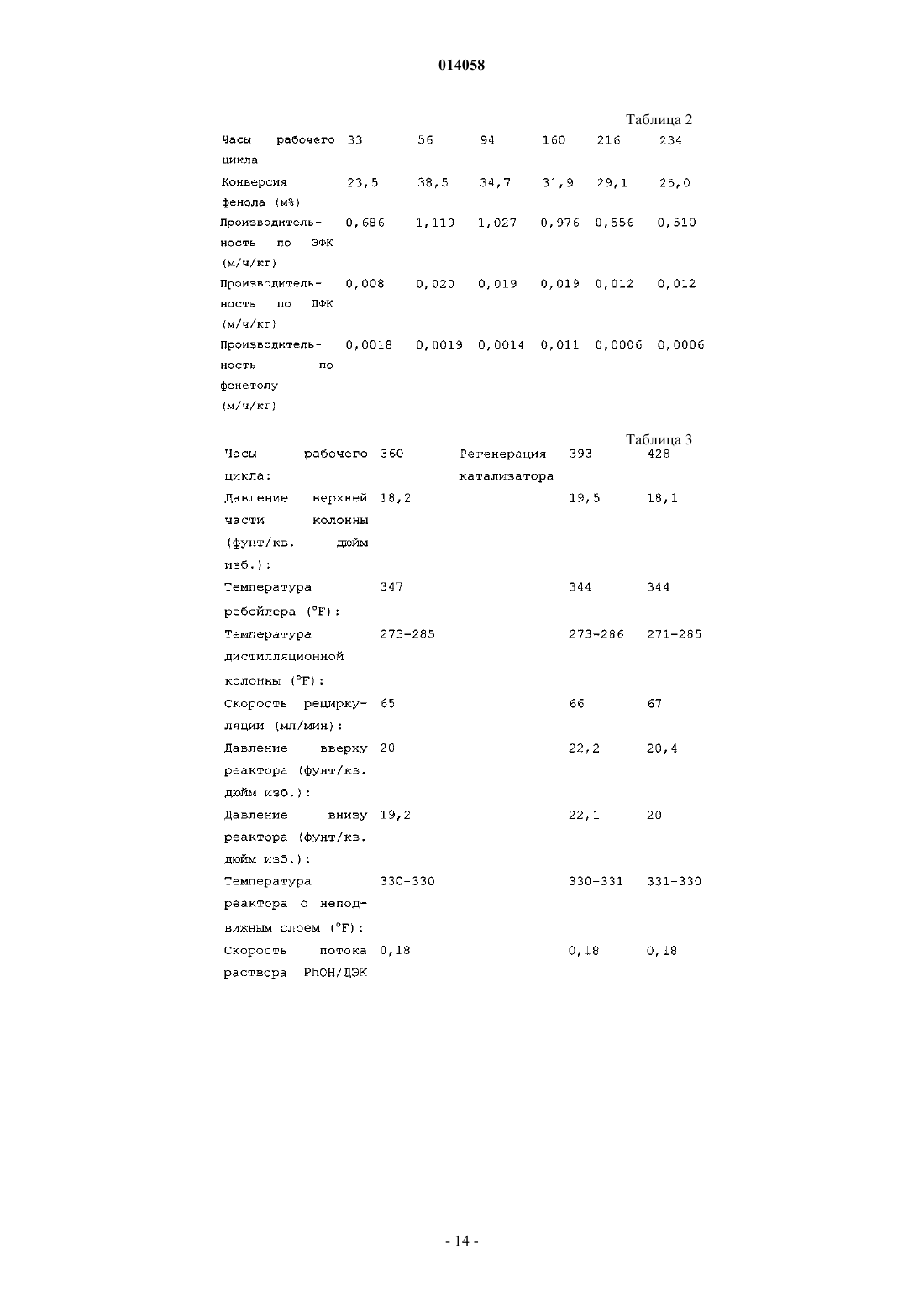
(71)(73) Заявитель и патентовладелец: КАТАЛИТИК ДИСТИЛЛЕЙШН ТЕКНОЛОДЖИЗ (US) Предложен способ получения различных органических карбонатов выполнением реакций переэтерификации и диспропорционирования в двухфазном пар/жидкость режиме, предпочтительно в присутствии композиции твердого катализатора, выбранного из группы, состоящей из оксидов,гидроксидов, оксигидроксидов или алкоксидов от двух до четырех элементов из групп IV, V и VI Периодической таблицы, нанесенного на пористый материал, который имеет поверхностные гидроксильные группы, и способ регенерации катализатора, дезактивированного осаждением полимера, контактированием дезактивированного катализатора с раствором гидроксисодержащего соединения в растворителе, таком как бензол или ТГФ. 014058 Уровень техники настоящего изобретения Область техники, к которой относится изобретение Настоящее изобретение относится к способу производства различных органических карбонатов проведением химических реакций, которые ограничиваются равновесием, и отделением различных химических соединений. Достижение более высоких конверсий, чем в условии равновесия, является весьма желательным для улучшения экономического аспекта производства. Настоящее изобретение раскрывает способ сдвига равновесия химической реакции для достижения более высокой конверсии. Диарилкарбонаты, например дифенилкарбонат (ДФК), являются важным сырьем для производства поликарбонатов. Современный уровень техники производит ДФК из диметилкарбоната (ДМК) и фенола в две стадии с использованием множества реакторов и гомогенного катализатора, такого как алкоксид титана. Следующие три реакции включены в производство ДФК. Равновесие каждой из приведенных выше реакций резко сдвинуто влево, в то время как цель настоящего изобретения состоит в том, что равновесие сдвигают вправо.J.L.R. Williams et al. (J. Org. Chem., 24 (1) pp. 64-68, 1959) обнаружили, что диспропорционирование несимметричных карбонатов, таких как метилфенилкарбонат (МФК), и симметричных карбонатов, таких как дибензилкарбонат, может быть выполнено в присутствии подходящего гомогенного катализатора, в частности алкоксида металла. Однако данная реакция сопровождается рядом нежелательных побочных реакций, таких как разложение соединений карбонатов до диоксида углерода, полимеризацией, образованием олефинов и эфиров. Авторы считают, что маршрут реакций зависит от структуры карбонатов и от катализатора, и что более щелочные катализаторы промотируют больше побочных реакций диспропорционирования алкилфенилкарбонатов, чем умеренно кислый катализатор, такой как бутоксид титана. Алкилфенилэфир является основным нежелательным побочным продуктом. Патент США 4045464 (1977) описывает способ получения диарилкарбонатов из фенилалкилкарбонатов через реакцию (3) в присутствии гомогенных льюисовских кислотных катализаторов с формуламиAlX3, TiX3, UX4, TiX4, VOX3, VX5 и SnX4, где Х является галогеном, ацетокси, алкокси или арилоксигруппой. ДФК получали проведением диспропорционирования этилфенила (ЭФК) или МФК при 180 С с 95% селективностью в присутствии гомогенных титановых катализаторов. Патент США 4554110 (1985) описывает улучшенный способ получения ароматических карбонатов из диалкилкарбоната и фенола в присутствии катализатора, включающего полимерные соединения олова. Диарилкарбонаты получают проведением диспропорционирования алкиларилкарбоната. Патент США 5210268 (1993) описывает способ получения диарилкарбонатов переэтерификацией в двух реакционных зонах. Смесь ароматических карбонатов получают переэтерификацией между диалкилкарбонатом, алкиларилкарбонатом и их смесью, и ароматическим гидроксисоединением на первой стадии. Диарилкарбонат получают в основном проведением диспропорционирования алкиларилкарбоната во второй стадии. Данный патент описывает способ преодоления неблагоприятного равновесия. Переэтерификацию выполняют в режиме реакционной дистилляции подачей реагентов в непрерывные многоступенчатые дистилляционные колонны в присутствии гомогенного или твердого катализатора, при непрерывном отводе полученной смеси ароматических карбонатов в качестве высококипящего продукта из нижней части дистилляционной колонны, и при непрерывном отводе легких побочных продуктов, таких как алифатический спирт или диалкилкарбонат, в виде потока пара из верхней части дистилляционных колонн. Диспропорционирование алкиларилкарбоната до диарилкарбоната и диалкилкарбоната также выполняют подобным образом. Диарилкарбонат непрерывно получают выполнением последовательно переэтерификации и диспропорционирования, используя множество многоступенчатых дистилляционных колонн. Патенты США 5872275 (1999) и 6262210 (2001) описывают способ получения диарилкарбоната из диалкилкарбоната и ароматического гидроксисоединения в присутствии жидкого гомогенного катализатора и способы удаления тяжелых высококипящих побочных продуктов и регенерации катализатора для повторного цикла. Патент США 6093842 (2000) описывает способ получения диарилкарбоната из диалкилкарбоната,ароматического гидроксисоединения и смешанного раствора, содержащего алкиларилкарбонат, введени-1 014058 ем трех потоков реагентов в экстракционную/реакционную дистилляционную колонну в присутствии катализатора. Примерами катализаторов являются соединения свинца, соединения меди, соединения щелочного металла, соединения никеля, соединения циркония, соединения титана, соединения ванадия и т.д. Побочными продуктами являются CO2, анизол, бензоаты и тяжелые материалы. Патент США 5426207 (1995) описывает способ получения диарилкарбоната, такого как ДФК, проведением переэтерификации ДМК с ароматическим гидроксисоединением и диспропорционирования алкиларилкарбоната в присутствии гомогенного катализатора в трех последовательных реакционных зонах. Условия выбирают, чтобы довести до максимума образование алкиларилкарбоната в первой и второй реакционных зонах, при этом диспропорционирование поддерживают в третьей реакционной зоне. Патент США 6767517 (2004) описывает способ непрерывного получения диарилкарбонатов. Данный способ использует реакторы реакционной дистилляции колонного типа и две ректификационные колонны для разделения промежуточного продукта реакции и конечного продукта. Чтобы облегчить недостатки, связанные с использованием гомогенных катализаторов для производства диарилкарбонатов, патенты США 5354923 (1994), 5565605 (1996) и WO 03/066569 (2003) описывают гетерогенные катализаторы.WO 03/066569 описывает способ непрерывного производства ароматического карбоната, такого как ДФК, в присутствии гетерогенного катализатора, полученного нанесением оксида титана на кремнеземJP 79125617 (1979) и JP заявка NO HEI 07 [1995]-6682 описывают гетерогенные катализаторы для получения дифенилкарбоната переэтерификацией ДМК с фенолом до МФК и диспропорционирование МФК до ДФК в присутствии MoO3 или V2O5, нанесенных на неорганический носитель, такой как кремнезем, оксид циркония или титана. Переэтерификацию и диспропорционирование проводят в реакторе дистилляционной башне, состоящей из реактора и дистилляционной башни с удалением побочных продуктов дистилляцией. Публикация Z.-H. Fu et al. (J. Mol. Catal. A: Chemical 118, (1997) p. 293-299) описывает синтез дифенилкарбоната из ДМК и фенола в присутствии различных гетерогенных металлоксидных катализаторов. Наилучшая селективность сообщается для MoO3 (20 мас.% оптимальная концентрация) катализатора,нанесенного на кремнезем. Из-за многих недостатков современных ДФК процессов, улучшенный процесс является весьма желательным для сбережения материалов, более дешевых затрат на сооружения, потребления меньшей энергии и затрат на эксплуатацию установки. Хотя предшествующий уровень техники не упоминает срок службы катализатора, все гетерогенные катализаторы со временем дезактивируются и становятся бесполезными. Гетерогенные катализаторы можно использовать коммерчески, только если их цикл и сроки службы являются достаточно долгими, или если данные катализаторы могут быть регенерированы in situ без серьезных финансовых затрат. Таким образом, проблемы дезактивации катализатора и метод регенерации остаются в качестве главных препятствий для коммерческого использования в предшествующем уровне техники. Сущность изобретения Данное изобретение относится к способу получения различных органических карбонатов путем проведения реакций переэтерификации и диспропорционирования в присутствии твердого катализатора в двухфазном реакторе. Настоящий способ является полезным для проведения химических реакций, ограниченных равновесием, включающих органические карбонаты, где как газовая фаза, так и жидкая фаза должны сосуществовать, чтобы сместить положение равновесия в правую сторону равновесной реакции,таким образом, приводя к высокой конверсии. Данные химические реакции проводят в присутствии одного или множества гетерогенных катализаторов. Предпочтительными твердыми катализаторами, описанными в данном изобретении, являются смешанные оксидные катализаторы, содержащие от двух до четырех различных элементов из группы IV, V и VI Периодической таблицы, предпочтительно Ti, Zr, Hf, Nb, Ta, Mo, V, Bi и Si, нанесенные на пористые материалы, такие как кремнезем, которые имеют поверхностные гидроксильные группы. Нанесенные металлические алкоксиды или смешанные металл-алкоксидные катализаторы групп IV и V металлические алкоксиды, такие как алкоксиды титана, алкоксиды циркония, алкоксиды ванадия, алкоксиды ниобия, VO(OR)3 или олигомеры оксоалкоксидов, и подобные составляют предпочтительную группу катализаторов. Катализаторы переэтерификации и диспропорционирования, используемые в настоящем процессе, могут быть одинаковыми или различными. В общем, гетерогенные катализаторы являются более желательными в сравнении с гомогенным катализатором из-за трудностей, связанных с повторным использованием гомогенных катализаторов. Однако все гетерогенные катализаторы со временем дезактивируются. Дезактивированные катализаторы должны быть заменены или свежим катализатором, или регенерированным in situ без особых трудностей. Для целей настоящего изобретения термин "двухфазный режим" означает любой процесс, имеющий как жидкую, так и паровую фазу, присутствующую в реакционной зоне, не взирая на средства достижения паровой и жидкой фазы, включая "реакционную дистилляцию", "каталитическую дистилля-2 014058 цию", кипение и конкурентную реакцию, и фракционную дистилляцию в колонне. Термин "обработанный носитель" или "обработанный кремнезем" означает носитель, имеющий оптимальное распределение поверхностных гидроксильных групп для данной площади поверхности для получения катализаторов,описанных здесь. В предпочтительном варианте осуществления способ получения диарилкарбоната включает:(b) подвод к указанной первичной реакционной зоне диалкилкарбоната и ароматического гидроксисоединения;(c) поддержание первичной реакционной зоны при двух фазах и реакционных условиях, способствующих образованию алкиларилкарбоната;(d) переэтерификацию данного диалкилкарбоната с ароматическим гидроксисоединением в присутствии твердого катализатора, выбранного из группы, включающей от двух до четырех элементов группIV, V и VI Периодической таблицы, нанесенных на пористый материал, который имеет поверхностные гидроксильные группы;(e) извлечение потока двухфазного продукта из первичной реакционной зоны;(f) отделение потока двухфазного продукта из (е) для извлечения парообразного алкилового спирта и жидкого алкиларилкарбоната;(g) поддержание вторичной реакционной зоны при двух фазах и реакционных условиях, способствующих диспропорционированию алкиларилкарбоната до диарилкарбоната;(h) диспропорционирование алкиларилкарбоната в присутствии твердого катализатора, выбранного из группы, включающей от двух до четырех элементов групп IV, V и VI Периодической таблицы, нанесенных на пористый материал, который имеет поверхностные гидроксильные группы;(i) извлечение потока двухфазного продукта из вторичной реакционной зоны; и(j) отделение потока двухфазного продукта из (i) для извлечения парообразного компонента, включающего алкиларилкарбонат, и жидкого продукта, включающего диарилкарбонат. Более предпочтительно проводят дополнительные стадии:(k) отделение диарилкарбоната дистилляцией и(l) возвращение арилалкилкарбоната во вторичную реакционную зону. Важно, чтобы носитель имел поверхностные гидроксильные группы. Кремнезем является предпочтительным носителем. Предпочтительно, чтобы носитель являлся обработанным носителем. Краткое описание чертежа Данный чертеж является схематическим изображением одного варианта осуществления способа по настоящему изобретению. Подробное описание Улучшения способа были сделаны введением двухфазных реакторов для проведения переэтерификации и диспропорционирования, чтобы установить следующие условия процесса получения ДФК:(2) снижение до минимума ДЭК в потоке подачи в реактор диспропорционирования;(3) проведение переэтерификации в присутствии фенола, так чтобы мольное отношение фенола к ЭФК было больше 0,2, предпочтительно больше примерно 0,3, наиболее предпочтительно больше примерно 0,35;(4) проведение диспропорционирования в присутствии фенола, при мольном отношении фенола к ДФК в диапазоне от 0,05 до 10, предпочтительно от 0,1 до 6, и мольном отношении фенола к ЭФК от 0,01 до 6;(5) необязательно введение следовых количеств воды в каталитическую реакционную зону диспропорционирования в количестве до 0,3 мас.%, предпочтительно до 0,1 мас.%;(6) извлечение выгоды из отсутствия азеотропа ДЭК/этанол, для отделения ДЭК от этанола;(7) по существу, устранение непрогретых мест в каталитических реакционных зонах;(8) устранение рецикла катализатора, отделения катализатора и непрерывного добавления добавочного катализатора;(9) эффективное перемешивание реагентов и продуктов и(10) предпочтительно поддержание присутствия как паровой, так и жидкой фаз в реакционных зонах, чтобы удалять низкокипящие продукты реакции в газовую фазу, получая высокую конверсию, не лимитированную константой равновесия. Реакторы с неподвижным слоем работают в двухфазном режиме, что означает сосуществование как пара, так и жидкости в реакционных зонах, создавая желаемые условия кипения в каталитической реакционной зоне двухфазного реактора. Тем не менее, работа двухфазного реактора в условии кипения не является необходимой, пока обе фазы газа и жидкости сосуществуют в реакционной зоне. Направление потока в двухфазном реакторе может быть или нисходящим, или восходящим. Нисходящий поток является предпочтительным. Также двухфазные реакторы с неподвижным слоем имеют петли рециркуляции. Реактор с кипящим нисходящим потоком работает, предпочтительно, так, чтобы-3 014058 иметь отрицательный перепад давления, что означает более низкое давление на дне слоя катализатора,по сравнению с верхом слоя катализатора. Отрицательный перепад давления создается высокой массовой скоростью потока парожидкостной смеси через неподвижный слой гетерогенного катализатора в каталитической реакционной зоне. Отрицательный перепад давления является желательным, но не необходимым. Перепад давления приблизительно 0,2 фунт/кв.дюйм на фут или более является предпочтительным. Работа двухфазного реактора в условиях точки кипения имеет несколько преимуществ перед работой традиционного реактора с неподвижным слоем, а именно массоперенос реагентов из объемной фазы в частицы катализатора и транспорт продуктов реакции из внутренних пор гранул катализатора, имеющих форму, в каталитическую реакционную зону, латеральное перемешивание реагентов и продуктов в объемной фазе, отсутствие роста или отрицательное падение температуры или место перегрева в каталитической реакционной зоне как для эндо-, так и для экзотермических реакций, удаление легких продуктов реакции из жидкой реакционной среды в газовую фазу, что приводит к благоприятному смещению равновесия. При заданной температуре реакции и скорости потока в каталитической реакционной зоне условие кипящей реакции в каталитической реакционной зоне создается контролем давления, которое определяется составом реакционной среды. Необязательно можно выбрать использование более низкокипящего растворителя, чтобы создать лучшие условия кипения в каталитической реакционной зоне, когда точка кипения реагента или продукта близка или выше, чем предполагаемая температура реакции. Данное изобретение является особенно полезным для реакций, где точка кипения продукта реакции выше, чем температура реакции, или очень близка к температуре реакции, так чтобы не было или был незначительный объем газовой фазы в реакционной зоне в условиях реакции. Примеры таких реакций включают получение дифенилкарбоната диспропорционированием этилфенилкарбоната или метилфенилкарбоната или получение бис-(2-этил-1-гексил)карбоната реакцией переэтерификации алкилкарбоната с 2-этил-1-гексанолом. Предпочтительная температура реакции составляет примерно от 200 до 400F. Поскольку ДМК и ДЭК кипят при температурах выше чем примерно 305F, создание паровой фазы в большом промышленном реакторе с неподвижным слоем становится трудным при относительно высоких скоростях потока жидкости. Регулируя поток объема пара к реактору при данной скорости потока жидкой реакционной смеси в двухфазном реакторе, как описано в данном изобретении, становится относительно легко создавать соответствующий объем газовой фазы в реакционной зоне при данных условиях реакции и поддержание устойчивого состояния работы реактора. Значительными преимуществами, полученными по отношению к предыдущему уровню техники,являются:(a) высокая производительность по целевым продуктам,(b) высокая селективность по целевым продуктам,(c) отсутствие отделения катализатора от потока продуктов реакции,(d) превосходная продолжительность работы катализатора,(e) меньшее потребление энергии и(f) легкая регенерация дезактивированного катализатора. Сырье для реактора переэтерификации предпочтительно включает свежий фенол и ДЭК и смешанный поток рецикла, содержащий ДЭК, PhOH и ЭФК. По существу, не содержащий этанол поток сырья в реактор переэтерификации создает благоприятные условия для превращения фенола и высокой скорости реакции. Это также верно для, по существу, не содержащего ДЭК потока сырья в реактор диспропорционирования. В каталитических реакционных зонах практически не существует непрогретых мест, что является важным для поддержания устойчивых скоростей реакции. Поскольку существуют как паровая,так и жидкая фазы в реакционных зонах переэтерификации и диспропорционирования в двухфазных реакторах, легкие продукты реакции (этанол и ДЭК в каждой реакционной зоне) испаряются в газовую фазу, что создает благоприятные условия для высокой производительности по намеченным продуктам. Поскольку реагенты или продукты не подвергаются воздействию излишне высокой температуры в присутствии катализатора, существует меньше нежелательных побочных продуктов, что облегчает и удешевляет очистку неочищенного потока продукта ДФК. Поскольку поток сырья в реакционную зону диспропорционирования может состоять из довольно высококипящих соединений, таких как ЭФК или МФК,азот, низкокипящий компонент или оба вводятся в реакционную зону, чтобы создать значительный объем газовой фазы в реакционной зоне. Примерами таких низкокипящих компонентов являются этиловый эфир, пропиловый эфир, диметиловый эфир, метан, этан, пропан, бутан, гексан, гептаны, толуол и ксилолы. Предпочтительно все или, по меньшей мере, существенную часть низкокипящих компонентов вводят в реакционную зону в виде перегретого пара. Поскольку этанол и ДЭК не образуют азеотроп, отделение ДЭК для рецикла приводит к меньшему потреблению энергии и меньшим капитальным затратам в сравнении с существующими промышленными процессами, где ДФК получают из ДМК и фенола. Существуют две или три реакционные зоны. В первой реакционной зоне алкиларилкарбонат, такой как ЭФК, получают переэтерификацией ДЭК с фенолом. Во второй реакционной зоне ДФК получают-4 014058 проведением диспропорционирования ЭФК. Данные реакционные зоны включают два реактора с неподвижным слоем. Необязательно три реакционные зоны могут включать три реактора с неподвижным слоем или два реактора с неподвижным слоем и каталитический дистилляционный колонный реактор. Данные реакторы загружают одним или двумя различными гетерогенными катализаторами. Примерами органических карбонатов, получаемых настоящим изобретением, являются ДФК, ЭФК,МФК, ДЭК, ДМК, бис-(2-этилгексил)карбонат и моноглицеридкарбонаты жирной кислоты. Настоящее изобретение особенно полезно в производстве дифенилкарбоната (ДФК) из ДЭК и фенола. Хотя ДЭК является предпочтительным диалкилкарбонатом для получения ДФК, понятно, что данный процесс также полезен для получения ДФК использованием ДМК или другого алкилкарбоната или алкиларилкарбоната. Чтобы избежать избыточного нагревания ребойлера при проведении переэтерификации и диспропорционирования в каталитическом дистилляционном реакторе колонного типа, низкокипящий компонент или смесь низкокипящих компонентов может быть закачана напрямую в ребойлер. Следовательно,данная методика является частью настоящего изобретения для получения различных органических карбонатов, как для твердых катализаторов, так и для гомогенного катализатора, используемых в процессе. Данная методика не была описана в предшествующем уровне техники для производства органических карбонатов, таких как ДФК, ДЭМ и ДЭК. Предпочтительными гетерогенными катализаторами являются нанесенные смешанные оксиды,гидроксиды, оксигидроксиды и алкоксиды элементов IV, V и VI групп, которые наносят на пористые носители. Смешанные оксидные катализаторы могут быть комбинациями двух, трех или четырех элементов, выбранных из Мо, Nb, Ti, V, Zr, Bi и Si. Данные элементы наносят в форме оксидов или гидроксидов или оксигидроксидов на пористый носитель, такой как кремнезем, оксид циркония и оксид титана. Носители могут быть таблетками, гранулами, экструдатами, сферами и подобным, с размерами примерно от 1 до 5 мм. Нанесение может быть выполнено в одну стадию или множеством стадий. Примерами смешанных оксидных катализаторов являются Nb2O3-TiO2, V2O3-TiO2, MoO3-TiO2, TiO2-ZrO2, Nb2O5V2O3, MoO3-V2O5, MoO3-ZrO2, TiO2-ZrO2-SiO2, TiO2-Nb2O5-SiO2, MoO3-Nb2O5-TiO2, V2O5-Nb2O5-TiO2,MoO3-Nb2O5-SiO2, TiO2-Bi2O3-SiO2, MoO3-Nb2O5-ZrO2, TiO2-Nb2O5-Bi2O3, MoO3-V2O5-TiO2, TiO2-Bi2O3SiO2, MoO3-Bi2O3-SiO2, TiO2-ZrO2-Bi2O3-SiO2 и TiO2-ZrO2-Nb2O5-Bi2O3-SiO2. Основной процедурой для получения данных смешанных оксидных катализаторов являются пропитка и соосаждение или комбинация данных двух методов, которые выполняют в одну стадию или множеством стадий. Можно выполнить пропитку пористого носителя одним, двумя или тремя металлическими компонентами или смешанный оксидный носитель приготовить соосаждением. Пропитку можно выполнять в одну стадию или множеством стадий. Продукты соосаждения или продукты пропитки, полученные в порошкообразных формах, подвергают подходящей термообработке при температурах примерно от 150 до 600 С. Порошкообразные материалы формуют до подходящего размера, равного примерно от 1 до 5 мм, для реактора с неподвижным слоем. Сформованные материалы прокаливают при температуре от 200 до примерно 750 С, предпочтительно примерно от 250 до 600 С на воздухе. Необязательно один или два металлических компонента могут быть нанесены на сформованный материал, приготовленный методом соосаждения или пропиткой или в форме порошка, или имеющего определенную форму, и затем прокалены при температуре от 200 до примерно 750 С, предпочтительно примерно от 250 до 600 С на воздухе. Соосаждение и пропитка могут быть выполнены из водной или органической фазы, такой как углеводороды, эфиры, кетоны,спирты и их смеси. Когда соосаждение выполняют из органической фазы, предпочтительно используют металлорганические соединения. Например, два различных раствора различных металлорганических соединений добавляют к подходящему органическому растворителю одновременно при энергичном перемешивании в условиях осаждения при подходящих температурах. Иногда при добавлении или после необходим третий раствор, чтобы вызвать гелеобразование или осаждение. Примером третьего раствора является вода,щелочной или кислый водный раствор в подходящем органическом растворителе, таком как спирт, эфир,кетон, органический эфир или их смеси. Другим необязательным методом является одновременное добавление первого металлорганического раствора и третьего раствора ко второму металлорганическому раствору при энергичном перемешивании. При необходимости осадки состаривают при подходящей температуре примерно от 25 до 200 С в течение от 30 мин до примерно 30 ч в подходящей среде. Иногда соосажденный продукт в водной среде состаривают в нейтральной, умеренно кислой или щелочной органической среде. Состаривающая среда может содержать или может не содержать небольшое количество воды, в зависимости от природы материала, который нужно состарить. Состаривающая среда может быть умеренно кислой или умеренно щелочной либо нейтральной. Состаренный продукт сушат при температуре примерно от 100 до 400 С и затем прокаливают при температуре примерно от 250 до 750 С. При необходимости пропитку одним или двумя элементами подходящего носителя выполняют, используя органический раствор, содержащий один или два металлорганических соединения или водный раствор, содержащий один или два соединения. Необязательно можно провести многократные пропитки, используя раз-5 014058 личные растворы. Однако можно выбрать для использования любой гетерогенный катализатор, описанный в предшествующем уровне техники, пока данный катализатор удобен для реактора с неподвижным слоем для работы большого промышленного реактора. Примерами гетерогенных катализаторов, описанных в предшествующем уровне техники, являются оксид титана, TS-1, Ti-MCM-41, оксид молибдена, оксид ванадия, оксид ниобия, оксид свинца и MgLa смешанный оксид в зависимости от ситуации и предпочтительно нанесенный, как описано в данном изобретении. Важно, чтобы носитель имел поверхностные гидроксильные группы. Кремнезем является предпочтительным носителем. Термин "обработанный носитель" или "обработанный кремнезем" следует понимать, как обозначающий носитель, имеющий оптимальное распределение поверхностных гидроксильных групп для данной площади поверхности, для приготовления катализаторов, описанных здесь. В зависимости от того, как приготовлен кремнезем, кремнезем может не иметь достаточного числа поверхностных гидроксильных групп для данной площади поверхности. Для такого кремнезема, кремнезем обрабатывают, чтобы ввести дополнительные поверхностные гидроксильные группы, водным щелочным раствором и затем тщательно промывают водой, за чем следует прокаливание при температуре от 280 до 650 С, перед использованием. Необязательно можно попробовать повторно гидратировать коммерчески доступный кремнеземный носитель. Повторно гидратированный кремнезем прокаливают при температуре от 280 до 650 С, чтобы оптимизировать плотность распределения поверхностных гидроксильных групп перед использованием. Следовательно, предпочтительным кремнеземным носителем, используемым для получения твердого катализатора, является "обработанный кремнезем". Классом предпочтительных носителей, в частности кремнеземных носителей, являются те, которые имеют поверхностные гидроксильные группы, увеличивающиеся при обработке щелочным раствором, как описано, чтобы получить максимальное число гидроксильных групп без разрушения физической целостности и прочности носителя. Регулирование содержания натрия на кремнеземном носителе является очень важным для получения ароматических карбонатов, таких как ЭФК и ДФК, поскольку такие щелочные примеси, как оксид щелочного металла, на кремнеземе вызывают нежелательные побочные реакции и ведут к нестабильности катализатора. Щелочной металл на кремнеземном носителе вызывает нестабильность работы катализатора и нежелательные побочные реакции для переэтерификации и диспропорционирования, которые производят алкиларилкарбонат и диарилкарбонат. Предпочтительный "обработанный кремнезем" будет иметь меньше чем примерно 0,05 мас.% Na, предпочтительно меньше чем примерно 0,03 мас.%Na. Обработка кремнезема водным раствором щелочного металла имеет дополнительное преимущество уширения пор. Однако выщелачивание слишком большого количества кремнезема из кремнеземного носителя в процессе обработки раствором щелочного металла может приводить к проблеме сохранения физической целостности и прочности. Другие катализаторы, описанные в данном изобретении, являются нанесенными металлическими алкоксидами или смешанными металлическими алкоксидными катализаторами, которые получают связыванием металлических алкоксидов с пористыми материалами носителей через кислородные мостиковые связи. Пористые материалы носителей должны иметь поверхностные гидроксильные группы, которые реагируют с алкоксигруппами с образованием кислородных мостиковых связей. Предпочтительным носителем является обработанный кремнезем, который имеет меньше чем примерно 0,05 мас.% Na,предпочтительно меньше чем примерно 0,03 мас.% Na. Оптимизация распределения поверхностных гидроксильных групп на кремнеземном носителе является очень важной, чтобы создать стабильные, прочно связанные металлические алкоксидные активные центры, поскольку высокая температура прокаливаний не участвует в связывании активных металлических атомов с поверхностью кремнезема через M-O-Si мостиковые связи. Увеличение до максимума числа M-O-Si мостиковых связей является весьма желательным. Таким образом, оптимальное распределение гидроксильных групп для данной поверхности включает максимальное число гидроксигрупп, которые могут быть получены для данной площади поверхности в пределах ограничений содержания щелочного металла и прочности носителя, как описано выше. Предпочтительными металлическими алкоксидами являются алкоксиды металлов IV и V групп, такие как алкоксиды титана, алкоксиды циркония, алкоксиды ванадия, алкоксиды ниобия и аналогичные. Алкоксиды металлов V группы включают алкоксид низшей валентности, такой как тетраалкоксид и окситриалкоксид, такой как VO(OR)3 или олигомеры оксоалкоксида. Примерами предпочтительных носителей являются кремнезем, оксид циркония, оксид титана, оксид титана-кремнезем, кремнезем-оксид алюминия и кремнезем-оксид циркония. Использование обработанного кремнезема является особенно важным для получения нанесенных металл-алкоксидных катализаторов. Алкоксидный катализатор на носителе может иметь один или два различных металлических алкоксида. Гетерогенные металл-алкоксидные катализаторы получают контактированием раствора металлического алкоксида или смешанного раствора двух различных металлических алкоксидов с носителем, таким как кремнезем при температуре примерно от 20 до 400F, предпочтительно примерно от 40 до 300F. Раствор алкоксида получают растворением одного или двух различных металлических алкоксидов в растворителе. Растворитель должен не мешать реакции образования кислородного мостика каким-либо об-6 014058 разом. Примерами таких растворителей являются углеводороды, эфиры, кетоны, спирты и их смеси. Когда два алкоксида различных металлов наносят на носитель, необязательно получают два раствора алкоксидов различных металлов и осуществляют их взаимодействие с носителем последовательно.M(OR)n+xOH (на поверхности носителя)(RO)n-x(O-)x (на катализаторе)+xROH,где n=4 или 5, х=1, 2, 3 или 4 и R=алкильная или арильная группа. Нанесенный титановый алкоксид является кислотным катализатором. Более высокая активность нанесенного титанового алкоксидного катализатора на кремнеземе в сравнении с титановым алкоксидом в гомогенном катализаторе относится к более высокой кислотности нанесенного Ti+4. Кислотность катализатора играет важную роль в катализируемых кислотой реакциях переэтерификации и диспропорционирования в производстве ароматических карбонатов. Можно приготовить нанесенный металл-алкоксидный катализатор in situ в качестве необязательного метода. Реакторы загружают обработанным носителем. Растворы металлических алкоксидов циркулируют через реактор при температуре от окружающей среды до примерно 400F. После образования нанесенного металл-алкоксидного катализатора любой оставшийся растворитель выпускают из реакторов. После промывания реакторов подходящим растворителем, таким как этанол, пентан или толуол, и необязательной термообработки катализаторов в токе инертного газа, такого как азот, при температуре примерно от 80 до 400F, предпочтительно примерно от 100 до 350F, катализаторы готовы для переэтерификации и диспропорционирования. Для получения нанесенных металлических или смешанных металлалкоксидных катализаторов, особенно важно использовать "обработанный носитель". Примером обработанного носителя является обработанный кремнезем, описанный выше. В настоящем изобретении наблюдали дезактивацию гетерогенных катализаторов. Это особенно верно для реакции диспропорционирования алкиларилкарбонатов, таких как МФК, ЭФК и т.д. Было обнаружено, что причиной дезактивации катализатора является отложение тяжелых полимеров на твердом катализаторе, которые блокируют активные центры катализатора и заполняют поры катализатора. Дезактивированный катализатор является или темно-коричневым, или черным, в зависимости от степени отложения полимера. Скорость дезактивации для данного гетерогенного катализатора намного выше для реакции диспропорционирования, чем для реакции переэтерификации. Было обнаружено, что дезактивированный катализатор может быть регенерирован in situ деполимеризацией полимеров, отложившихся на катализаторе, контактированием дезактивированного катализатора с соединением, содержащим гидроксильную группу, таким как пар, метанол, этанол, фенол или смеси гидроксисоединений при повышенной температуре. Необязательно можно использовать раствор гидроксисоединения в растворителе. Предпочтительными растворителями являются бензол, толуол, ксилолы, пентан, гексан, октан, декан,ТГФ или любые смеси растворителей. Использование растворителя для деполимеризации полимеров,отложившихся на дезактивированном катализаторе, является только необязательным и нетребующимся. Регенерированный катализатор предпочтительно сушат при температуре от 200 до примерно 500F в токе инертного газа (например, азота) перед использованием. Регенерацию катализатора гидроксисодержащими соединениями выполняли обработкой дезактивированных катализаторов in situ в потоке спирта или раствора вода-спирт или паром при температуре от 250 до 600F, предпочтительно от 270 до 450F. Обработка дезактивированных катализаторов может быть выполнена как спиртовым раствором, так и паром. Например, дезактивированный катализатор может быть обработан сначала спиртом или спиртовым раствором, и затем паром или в обратном порядке. Предпочтительно проводить регенерацию катализатора под достаточным давлением, чтобы, по меньшей мере, некоторое количество жидкой фазы присутствовало в слое катализатора. Предпочтительным спиртом является метанол, этанол или смесь этих двух спиртов. Необязательно спиртовой раствор может содержать воду в количестве до 80 мас.%, предпочтительно до 20 мас.%, наиболее предпочтительно до 5%. Поток из реактора деполимеризации содержит фенол, ДЭК, ЭФК и следовые количества фенетола и тяжелых соединений в качестве продуктов реакции деполимеризации. Чтобы использовать гликолевый эфир для смывания полимеров на катализаторе и улучшить селективность, растворитель, как описано выше, вводят в качестве компонента реакционной смеси в каталитическую реакционную зону и отделяют от реакционной смеси для извлечения. Примерами таких эфирных растворителей являются диэтиловый эфир этиленгликоля, диметиловый эфир этиленгликоля, диэтиловый эфир диэтиленгликоля, диметиловый эфир диэтиленгликоля и т.д. Методика регенерации катализатора, описанная в данном изобретении, может также быть использована в любом процессе производства ароматических карбонатов, где используют гомогенный катализатор. Для регенерации гомогенной каталитической системы спиртовой раствор должен быть должным образом осушен, так чтобы содержание воды не превышало примерно 0,2 мас.%. Следовательно, методика регенерации катализатора, описанная в данном изобретении, применима для любого способа получения органических карбонатов. Неожиданно было обнаружено, что данная дезактивация катализатора могла быть смягчена проведением реакции диспропорционирования в присутствии ароматического гидроксисоединения, такого как фенол. Более того, существует дополнительная польза при проведении диспропорционирования в присутствии следовых количеств воды в каталитической реакционной зоне в количестве до 0,3 мас.%, пред-7 014058 почтительно до 0,10 мас.%. Оказывается, что существенная часть тяжелых полимеров, отлагающихся на катализаторах, является поликарбонатами. Проведение диспропорционирования в присутствии ароматического гидроксисоединения или как ароматического гидроксисоединения, так и следовых количеств воды в каталитической реакционной зоне приводит к приемлемой стабильной работе катализатора. Однако понятно, что слишком большое количество ароматического гидроксисоединения приводит к неприемлемо низкой скорости производства ДФК из-за равновесной природы реакции переэтерификации. Следовательно, поддержание мольного отношения ароматического гидроксисоединения к ДФК от 0,05 до 10, предпочтительно от 0,1 до 6, в зоне реакции диспропорционирования является существенным для обеспечения как долгого приемлемого времени каталитического цикла, так и хорошей производительности по ДФК. Для реакции переэтерификации мольное отношение фенола к ДФК поддерживают больше 0,2, предпочтительно больше 0,3, наиболее предпочтительно больше примерно 0,35. Пример схемы технологического процесса для получения ДФК иллюстрируется на фигуре. Существуют два двухфазных реактора с неподвижным слоем и пять дистилляционных колонн. Первый двухфазный реактор 38 является первичным для реакции переэтерификации для производства ЭФК из ДЭК и фенола. Второй двухфазный реактор 37 является первичным для реакции диспропорционирования для производства ДФК из ЭФК. Оба двухфазных реактора 38 и 37 загружают или гетерогенным катализатором, или необязательно двумя различными гетерогенными катализаторами, описанными в данном изобретении. Необязательно можно ввести дополнительные реакторы с неподвижным слоем (не показаны) в ряду между 38 и 37. Основной целью данного дополнительного реактора является производство дополнительного ЭФК. Свежий поток сырья фенола 1 и свежий поток сырья ДЭК 2 смешивают с рециркулирующими потоками 11 и 25, и затем вводят в двухфазный реактор 38 через линию 3. Необязательно поток газообразного азота вводят в 38 через линию 4. Продукт ЭФК и побочный продукт этанол получают проведением переэтерификации в 38. В реакторе 38 побочный продукт этанол испаряют в газовую фазу. Выходящий поток 5 реактора вводят в первую дистилляционную колонну 30 через линии 5, 6 и 7. Этанол удаляют в колонне 30 в поток верхнего погона 8 с малым количеством ДЭК. Поток 8 частично охлаждают и затем вводят в барабан разделения газ-жидкость 36, а поток газа 9 рециркулирует в реактор 38 и 37. Поток жидкости 10 (этанол) из 36 рециркулирует в производство ДЭК для получения ДЭК. Боковой поток 11 из колонны 30, который состоит из ДЭК, фенола и ЭФК, рециркулирует обратно в реактор 38 через линию 3. Необязательно можно выбрать размещение слоя катализатора 101 в нижней секции колонны 30 ниже боковой точки для дополнительной конверсии фенола в ЭФК. Дистилляционная колонна 30 разработана и действует так, чтобы боковой поток 11, по существу, не содержал этанол. Петля рециркуляции для двухфазного реактора 38 включает линии 5, 6 и 7, колонну 30 и линии 11 и 3. Нижний поток 12 из колонны 30 вводят во второй двухфазный реактор 37 через линии 13 и 14. Поток 12 содержит ЭФК и фенол. Но он также содержит небольшие количества ДФК и побочного продукта фенетола. Колонна 30 также разработана и действует так, чтобы минимизировать ДЭК в нижнем потоке 12. Поток 12 объединяют с потоком рецикла 19 в поток 13. Поток 13 объединяют с потоком газообразного азота 15 в поток 14, который вводят в 37. Продукт ДФК и побочный продукт ДЭК получают в реакторе 37 проведением диспропорционирования ЭФК. ДЭК в реакторе 37 испаряют в газовую фазу. Выходящий поток 16 реактора вводят во вторую дистилляционную колонну 32. Поток 16 состоит в основном из ДЭК, ЭФК, фенола, ДФК и небольших количеств этанола и побочных продуктов. Этанол и ДЭК в потоке 16 удаляют с паром в колонне 32 в качестве потока верхнего погона 17, который вводят в первую колонну 30 через линию 7. Нижний поток 18 из колонны 32 разделяется на два потока 19 и 20. Поток 19 рециркулирует обратно во второй реактор 37 через линии 13 и 14. Петля рециркуляции для второго двухфазного реактора 37 включает линию 16, колонну 32 и линии 18, 19, 13 и 14. Другой поток 20 вводят в третью дистилляционную колонну 34, где оставшийся ДЭК в потоке извлекают в качестве потока верхнего погона 21,который направляют в первую колонну 30 через линии 6 и 7. Боковой поток 22 из 34 вводят в четвертую дистилляционную колонну 35, чтобы удалить побочный продукт фенетол в качестве потока верхнего погона 24. Нижний поток 25 из колонны 35 рециркулирует в 38. Необязательно нижний поток 25 из 35 может быть возвращен в первую колонну 30 через линии 31, 21, 5, 6 и 7. Нижний поток 23 из 34 вводят в дистилляционную колонну 33 для извлечения продукта ДФК. Поток верхнего погона 26, который в основном состоит из ЭФК, возвращают в реактор диспропорционирования 37 через линию 14. Колонна 33 работает под давлением ниже атмосферного. Нижний поток 27 из 33 является потоком неочищенного ДФК. Можно выбрать для использования другой материал, такой как диэтиловый эфир, диметиловый эфир, изопентан или бутан вместо газообразного азота или частично заменить газообразный азот для работы двух двухфазных реакторов 38 и 37. Контрольный пример 1. Катализатор, представляющий собой оксида титана (9,2 мас.%), нанесенный на кремнезем, готовят в соответствии с предшествующим уровнем техники (WO O3/066569). 3,839 Ti(OC4Hg-n)4 растворяют в 90 мл сухого толуола. Гранулированный кремнезем (+8 меш, 655 ч./млн Na по массе, 300 м 2/г БЭТ SA и 1 см 3/г PV) был предварительно высушен при 330 С в течение 2 ч на воздухе. Раствор бутоксида титана кипятили с обратным холодильником с 25 мл (9,159) высушенных гранул кремнезема в кипящем растворе толуола в 200 мл колбе с холодильником. После примерно 6 ч кипячения избыток толуола в колбе-8 014058 отделяли выпариванием из колбы. Бутоксид титана, нанесенный на кремнезем, извлекали из колбы и сушили при 120 С в вакуумном шкафу в течение 1,5 ч. Сухой кремнезем прокаливали при 500 С в течение 2 ч. Прокаленный продукт представлял собой гранулы чистого белого цвета. Общая масса катализатора составляла 9,67 г. Данный катализатор являлся катализатором А. Пример 2. Катализатор, представляющий собой смешанный оксид ниобия/титана, нанесенный на кремнезем,готовили в соответствии с настоящим изобретением. 0,592 г Nb(OC4H9-n)5 растворяли в 80 мл толуола. Гранулированный кремнезем, использованный в контрольном примере 1, сушили при 320 С в течение 2 ч на воздухе. 25 мл (9,27 г) данного высушенного кремнезема кипятили с обратным холодильником с вышеуказанным раствором бутоксида ниобия. После 5,5 ч кипячения таким же образом, как и в контрольном примере 1, избыток толуола вылили из колбы. Водный раствор в метаноле, полученный смешением 0,209 г воды со 120 мл метанола, наливали в колбу, и затем раствор кипятили с обратным холодильником в кипящем метаноле в течение часа. Избыток метанола был высушен из колбы. Раствор тетрабутоксида титана, полученный растворением 3,56 г бутоксида титана в 90 мл толуола, наливали в колбу и затем содержимое колбы кипятили с обратным холодильником в толуоле в течение 5,5 ч. Избыток толуола в колбе отделяли выпариванием из колбы. Вещество в колбе извлекали из колбы и сушили при 120 С в вакуумном шкафу в течение 1,5 ч. Высушенный кремнезем прокаливали при 500 С в течение 2 ч. Внешний вид прокаленного продукта был отличным от катализатора в контрольном примере 1. Он был больше похож на гранулированный кремнеземный носитель, чем катализатор чистого белого цвета в контрольном примере 1. Общая масса катализатора составляла 10,28 г. Данный катализатор являлся катализатором В. Пример 3. В данном примере обработанный кремнезем использовали для получения катализатора, представляющего собой смешанный оксид титана/ниобия, нанесенного на кремнезем. Катализатор, представляющий собой смешанный оксид титана/ниобия, нанесенный на обработанный кремнезем, готовили в соответствии с настоящим изобретением. Тот же самый гранулированный кремнезем контрольного примера 1 использовали для приготовления обработанного кремнезема данного примера. Гранулированный кремнезем (40,56 г) обрабатывали раствором гидроксида натрия, приготовленного растворением 8,059NaOH в 226 г воды при комнатной температуре в течение 7 мин при перемешивании. Обработанный кремнезем тщательно промывали холодной водой и затем горячей водой (примерно 65 С) несколько раз,чтобы удалить следы натрия на кремнеземе. Обработанный кремнезем сушили при 150 С в течение 2 ч и затем прокаливали при 325 С в течение 2 ч. Данный прокаленный кремнезем содержал 300 ч./млн Na по массе. Раствор алкоксида ниобия готовили растворением 0,844 г Nb(OC4H9-n)5 в 80 мл толуола. 8,46 г обработанного кремнезема кипятили с обратным холодильником в вышеуказанном растворе бутоксида ниобия в течение 3 ч в колбе с охлаждаемым водой холодильником. Смесь вода-метанол получали смешением 0,645 г воды с 90 мл метанола. Данную смесь вода-метанол наливали в колбу и содержимое в колбе снова кипятили с обратным холодильником. После часа кипячения избыток раствора из колбы сливали. Раствор тетрабутоксида титана, полученный растворением 3,67 г бутоксида титана в 80 мл толуола, наливали в колбу, и затем содержимое колбы кипятили с обратным холодильником в течение 1 ч 45 мин. Избыток толуола в колбе отделяли выпариванием из колбы. Вещество в колбе извлекали из колбы и сушили при 120 С в вакуумной печи в течение 1 часа. Высушенный кремнезем прокаливали при 500 С в течение 2 ч. Внешний вид данного катализатора был больше похож на гранулированный кремнеземный носитель. Данный катализатор являлся катализатором С. Пример 4. Катализатор, представляющий собой алкоксид титана, нанесенный на обработанный кремнезем,получали в соответствии с настоящим изобретением. Тот же самый гранулированный кремнезем(80,55 г), использованный в контрольном примере 1, обрабатывали раствором гидроксида натрия, приготовленного растворением 8,3 г NaOH в 580 мл деионизированной воды при комнатной температуре в течение 8 мин при перемешивании. Обработанный кремнезем промывали холодной водой и затем горячей водой. Промытый кремнезем обрабатывали раствором нитрата аммония, полученного растворением 99 г нитрата аммония в 2 л деионизированной воды при 80 С в течение 2 ч. Обработку кремнезема раствором нитрата аммония повторяли 13 раз. В конце кремнезем промывали деионизированной водой при температуре окружающей среды. Промытый кремнезем сушили при 110 С в течение часа, после чего следовало прокаливание при 370 в течение 1,5 ч и затем при 375 С в течение 30 мин. Прокаленный кремнезем содержал 23 ч./млн Na по массе и 2,9 мас.% потери массы при прокаливании при 550 С. Тетрабутоксид титана получали растворением 4,74 г тетрабутоксида титана в 70 мл толуола. 10,44 г (31 мл) обработанного кремнезема кипятили с обратным холодильником в растворе тетрабутоксида титана в течение 6 чв, и затем избыток раствора титана в колбе высушивали. Высушенный раствор титана содержал 0,50 мас.% Ti. Кремнезем промывали 90 мл толуола при комнатной температуре. Промытый продукт сушили при 170 С в течение 3 ч в вакуумном шкафу. Внешний вид конечного катализатора был похож на обработанные гранулы кремнезема. Небольшую часть конечного катализатора прокаливали при 500 С в течение 2 ч на воздухе, чтобы определить содержание Ti в данном катализаторе. Содержание Ti прока-9 014058 ленного катализатора составляет 3,25%. Внешний вид прокаленного катализатора был похож на обработанные гранулы кремнезема. Данный эксперимент показывает успешное удержание алкоксида титана на обработанном кремнеземе. Проведение переэтерификации и диспропорционирования. Катализаторы тестировали в устройстве, имеющем реактор с неподвижным слоем, дистилляционную колонну и сборник орошающей фракции. Размер реактора с неподвижным слоем составлял 1/2 дюйма в диаметре и 15 дюймов длиной. Он имел три термопары для наблюдения за температурой в трех положениях прямо над слоем катализатора, в середине слоя катализатора и прямо под слоем катализатора. Температуры верхней половины и нижней половины реактора контролируют независимо. Устройство также имеет подогреватель сырья наверху неподвижного слоя катализатора, температура которого контролируется отдельно. Реактор с неподвижным слоем работал в режиме нисходящего потока. Дистилляционная колонна состоит из ребойлера емкостью 2 л и колонны 42"1" внешний диаметр(0,870" внутренний диаметр). Имелись три датчика давления для контроля и записи давления вверху колонны, верха и низа слоя катализатора. Также ребойлер имел датчик уровня жидкости. Неподвижный слой имел петлю рециркуляции; поток в реактор из среды жидкости в ребойлере накачивали через реактор нисходящим потоком и затем возвращали в ребойлер. Свежее сырье ДЭК закачивали в петлю рециркуляции перед реактором. Свежий раствор исходного фенола отдельно закачивали в петлю рециркуляции перед реактором. Газообразный азот вводили в систему при необходимости. Пар из дистилляционной колонны конденсировали и удаляли в качестве потока верхнего погона жидкости. Неконденсируемый газ выгружали из холодильника. Для непрерывной работы переэтерификации раствор исходного фенола непрерывно подавали при определенной скорости, в то же время непрерывно удаляя поток продукта из ребойлера при заданной постоянной скорости и непрерывно удаляя верхний продукт. Скорость подачи сырья ДЭК является каскадной для поддержания постоянного уровня жидкости в ребойлере. Тест 1. Цель данного цикла состоит в демонстрации работы двухфазного реактора с неподвижным слоем в реакции переэтерификации. Двойная фаза в каталитической реакционной зоне создавалась кипением смеси паровой фазы и жидкой фазы в каталитической реакционной зоне. Катализатор А (25 мл; 9,67 г) загружали в реактор. В ребойлер загружали 167,6 г фенола и 737,4 г ДЭК. Испытание проводили при следующих условиях. Давление верхней части колонны: 18 фунт/кв.дюйм изб. Температура ребойлера: 335F. Температура дистилляционной колонны: 300-310F. Скорость рециркуляции: 66 мл/мин. Давление наверху реактора с неподвижным слоем: 24,6 фунт/кв.дюйм изб. Давление внизу реактора с неподвижным слоем: 20,5 фунт/кв.дюйм изб. Температура реактора с неподвижным слоем: 338-342F. Скорость потока азота в ребойлер: 60 мл/мин. 0 флегмы из сборника орошающей фракции. Отсчет времени работы начался, когда температура реактора с неподвижным слоем достигла заданной температуры 340F. В процессе работы ДЭК непрерывно закачивали для поддержания постоянного уровня жидкости. После работы в течение 45 ч, 23 г фенола загружали в систему в виде 20,92 мас.% раствора фенола в течение периода времени в 55 мин при скорости потока 2 мл/мин. Работу продолжали до 77 ч рабочего цикла. Средняя скорость потока верхнего погона жидкости составляла примерно 0,3 мл/мин. Поток верхнего погона состоял в основном из ДЭК и небольшого количества этанола. Образцы были взяты из ребойлера и потока верхнего погона для анализа. В конце 77 ч времени работы превратилось 64,8 мол.% фенола, загруженного в устройство. Выходы составили 63,1 мол.% ЭФК; 1,52 мол.% ДФК и 0,26 мол.% побочных продуктов, исходя из общего количества фенола, загруженного в систему. Фенетол был главным побочным продуктом, что отвечало 33,6 мол.% побочных продуктов. Средняя производительность составляла 1,81 м/ч/кг катализатора для ЭФК; 0,041 м/ч/кг катализатора для ДФК и 0,01 м/ч/кг катализатора для побочных продуктов. Производительность ЭФК в данном примере является лучшей по отношению к описанным в предшествующем уровне техники, демонстрируя лучшую производительность процесса, описанного в настоящем изобретении. Тест 2. Цель данного цикла состоит в демонстрации работы двухфазного реактора с неподвижным слоем и катализатора, представляющего собой смешанный оксид ниобия и титана в соответствии с настоящим изобретением для реакции переэтерификации. Двойную фазу в каталитической реакционной зоне создавали кипением смеси паровой фазы и жидкой фазы в каталитической реакционной зоне. Катализатор В (24 мл; 9,172 г) загружали в реактор. Данный тест выполняли в условиях, идентичных тесту 1. В ребойлер загружали 167,6 г фенола и 737,4 г ДЭК. Отсчет времени работы начался, когда температура реактора с неподвижным слоем достигла заданной температуры 340F. В процессе работы- 10014058 ДЭК непрерывно закачивали для поддержания постоянного уровня жидкости. После работы в течение 22 ч 91,62 г фенола загружали в систему в виде 31,38 мас.% раствора фенола в течение периода времени,равного 2 ч 20 мин, при скорости потока 2 мл/мин. Работу продолжали до 73 ч рабочего цикла. Средняя скорость потока жидкости верхнего погона составляла примерно 0,3 мл/мин. Поток верхнего погона состоял в основном из ДЭК и небольшого количества этанола. Образцы были взяты из ребойлера и потока верхнего погона для анализа. В конце 73 ч времени работы превратилось 64,6 мол.% фенола, загруженного в устройство. Выходы составили 63,1 мол.% ЭФК; 1,4 мол.% ДФК и 0,27 мол.% побочных продуктов, исходя из общего количества фенола, загруженного в систему. Фенетол был главным побочным продуктом, что отвечало 55,9 мол.% побочных продуктов. Средняя производительность составляла 2,567 м/ч/кг катализатора для ЭФК; 0,059 м/ч/кг катализатора для ДФК и 0,022 м/ч/кг катализатора для побочных продуктов. Основным побочным продуктом был фенетол, что отвечало 33,6 мол.% всех побочных продуктов. Производительность по ЭФК в данном примере является лучшей по отношению к катализатору, описанному в предшествующем уровне техники и тесте 1. Тест 3. Данный тест проводили для демонстрации диспропорционирования ЭФК в ДФК в периодическом процессе. Реактор с неподвижным слоем работал в режиме точки кипения с использованием такого же оборудования, как описано. Неочищенное сырье получали как в тесте 2, и затем избыток ДЭК в неочищенном сырье был отогнан, чтобы сконцентрировать ЭФК, и затем к нему добавляли толуол. Целью добавления толуола к концентрированному неочищенному сырью являлось проведение диспропорционирования в двухфазном режиме в реакторе с нисходящим потоком. Кипящую реакционную смесь паровой фазы и жидкой фазы создавали в каталитической реакционной зоне. Отсчет времени работы начался, когда температура реактора с неподвижным слоем достигла заданной температуры 340F. Тест проводили с тем же катализатором, использованным в тесте 2. В процессе работы толуол непрерывно закачивали для поддержания постоянного уровня жидкости и скорости потока верхнего погона примерно 7 мл/мин. Поток верхнего погона состоял в основном из толуола, небольшого количества ДЭК и следового количества этанола. Общая масса сырья в ребойлере составляла 635,3 г. Состав сырья содержал 44,19 мас.% толуола, 8,42 мас.% ДЭК; 0,07 мас.% фенетола; 0,29 мас.% побочных продуктов; 11,21 мас.% фенола; 31,88 мас.% ЭФК и 3,94 мас.% ДФК. Испытание проводили при следующих условиях. Давление верхней части колонны: 16,3 фунт/кв.дюйм изб. Температура ребойлера: 311F. Температура дистилляционной колонны: 295-305F. Скорость рециркуляции: 67 мл/мин. Давление наверху реактора с неподвижным слоем: 32,8 фунт/кв.дюйм изб. Давление внизу реактора с неподвижным слоем: 18,5 фунт/кв.дюйм изб. Температура реактора с неподвижным слоем: 327-330F. Скорость потока азота в ребойлер: 50 мл/мин. 0 флегмы из сборника орошающей фракции. После 6 ч работы анализ продукта из ребойлера показал 5,8 мол.% конверсии ЭФК с 95,2 мол.% селективности по ДФК. Производительность по ДФК составила 0,614 м/ч/кг катализатора. Тест 4. Данный тест проводили для демонстрации непрерывной работы для получения ЭФК в реакторе с неподвижным слоем, работавшем в режиме точки кипения в условиях установившегося режима. Была приготовлена другая партия катализатора, идентичного катализатору В в примере 2. 9,04 г(примерно 25 мл) данного катализатора загружали в реактор. 200 г фенола и 610 г ДЭК загружали в ребойлер для непрерывной работы. Отсчет времени работы начался, когда температура реактора с неподвижным слоем достигла заданной температуры 340F. В течение цикла ДЭК непрерывно закачивали для поддержания постоянного уровня жидкости. После работы в течение 23,25 часов, 85,58 г фенола загружали в систему в виде 31,38 мас.% раствора фенола в течение периода времени, равного 2 ч 15 мин, при скорости потока 2 мл/мин. Непрерывную работу в условиях установившегося режима проводили непрерывной закачкой 32,2 мас.% раствора фенола в ДЭК при 0,18 мл/мин и непрерывным удалением потока продукта при 0,15 мл/мин. ДЭК закачивали для поддержания постоянного уровня жидкости 81% в ребойлере. За 242,5 ч рабочего цикла в установившемся режиме были получены следующие результаты. Рабочий цикл: 242,5 ч. Давление верхней части колонны: 19,6 фунт/кв.дюйм изб. Температура ребойлера: 354F. Температура дистилляционной колонны: 330F. Скорость рециркуляции: 67 мл/мин. Давление наверху реактора с неподвижным слоем: 20,8 фунт/кв.дюйм изб. Давление внизу реактора с неподвижным: слоем: 19,2 фунт/кв.дюйм изб. Температура реактора с неподвижным слоем: 341F. Скорость потока азота в ребойлер: 60 мл/мин.- 11014058 Флегма из сборника орошающей фракции: 0. Уровень жидкости в ребойлере: 81,04%. Скорость подачи 32,2 мас.% PhOH/ДЭК раствора: 0,15 мл/мин. Скорость потока ДЭК: 0,28 мл/мин. Скорость потока верхнего погона: 0,269 мл/мин. Скорость потока нижнего продукта: 0,185 мл/мин. Состав потоков (мас.%): Конверсия фенола (мол.%): 39,5. Выход ЭФК: 37,6 мол.%. Селективность ЭФК: 95,04 мол.%. Выход ДФК: 1,9 мол.%. Селективность ДФК: 4,87 мол.%. Селективность фенетола: 0,05 мол.%. Производительность ЭФК: 1,50 м/ч/кг катализатора. Фенетол был единственным побочным продуктом, зарегистрированным в потоке продукта гх и гхмс. Данный результат является лучшим по отношению к предшествующему уровню техники. Наблюдалась дезактивация катализатора. Тест 5. Данный тест проводили для демонстрации непрерывной работы для реакции переэтерификации между пропиленкарбонатом и этанолом для получения ДЭК и побочных продуктов пропиленгликоля,используя реактор с неподвижным слоем, работавший в режиме точки кипения. Была приготовлена другая партия катализатора, идентичного катализатору В в примере 2. 9,6 г(примерно 25 мл) данного катализатора загружали в реактор. 280 г пропиленкарбоната и 635 г этанола загружали в ребойлер для непрерывной работы. Отсчет времени работы начался, когда температура реактора с неподвижным слоем достигла заданной температуры 335F. В течение цикла поток продукта непрерывно удаляли из ребойлера при постоянной скорости 0,14 мл/мин. Для поддержания постоянного уровня жидкости в 85%, 23,1 мас.% раствор пропиленкарбоната в этаноле непрерывно закачивали в реактор каскадного типа. Работа продолжалась 162 ч. В течение цикла конверсия пропиленкарбоната была довольно постоянной. Средняя конверсия пропилена в течение цикла составила 14,5 мол.%. Средняя производительность по ДЭК составила 1,81 моль/ч/кг катализатора. Тест 6. Цель данного эксперимента состоит в демонстрации лучшей работы смешанного оксидного катализатора, нанесенного на обработанный кремнезем, по отношению к традиционному катализатору, описанному в предшествующем уровне техники. Были продемонстрированы более медленная скорость дезактивации, более высокая активность катализатора С и регенерация катализатора деполимеризацией дезактивированного катализатора С. Испытание традиционного катализатора, представляющего собой оксид титана, нанесенного на кремнезем (контрольный пример 1), было проведено в тесте 6 А. Испытание смешанного Ni/Nb оксидного катализатора, нанесенного на обработанный кремнезем (катализатор С в примере 3), было выполнено в примере 6 В. Тест 6 А. Готовили другой Ti оксидный катализатор, подобный катализатору А в контрольном примере 1. Единственным отличием от контрольного примера 1 было прокаливание такого же кремнезема при 340 С в течение 4 ч на воздухе. 25 мл (9,0 г) катализатора загружали в описанный реактор с неподвижным слоем. Внешний вид катализатора представлял собой сахарно-белые покрытые гранулы кремнезема,такие же как катализатор в контрольном примере 1. 285 г фенола и 530 г ДЭК загружали в ребойлер. Испытание проводили при следующих условиях. Давление верхней части колонны: 19-21 фунт/кв.дюйм изб. Температура ребойлера: 345-347F. Температура дистилляционной колонны: 270-285F. Скорость рециркуляции: 66-67 мл/мин. Давление наверху реактора с неподвижным слоем: 20-22 фунт/кв.дюйм изб. Давление внизу реактора с неподвижным слоем: 19-22 фунт/кв.дюйм изб. Температура реактора с неподвижным слоем: 332-337F.- 12014058 Скорость потока азота в ребойлер: 60 мл/мин. 0 флегмы из сборника орошающей фракции. Скорость потока PhOH/ДЭК раствора: 0,17-0,18 мл/мин. Скорость нижнего потока: 0,18-0,19 мл/мин. В процессе работы поток ДЭК падал до уровня жидкости 88% (условная шкала) для поддержания постоянного уровня жидкости. Работу выполняли непрерывным сливом реакционной смеси в ребойлер, в то же время закачивая 40,4 мас.% раствора фенола в ДЭК при постоянной скорости, составляющей около 0,19 мл/мин. Цикл продолжали в течение 115 ч без перерыва. Поток верхнего погона состоял в основном из ДЭК, этанола и небольших количеств фенола и ЭФК. Образцы были взяты из ребойлера и потока верхнего погона для анализа. Фенетол был единственным продуктом в нижнем потоке продукта из ребойлера. Результаты собраны в табл. 1. Таблица 1 Тест 6 В. Работу катализатора С, полученного в примере 3, тестировали для переэтерификации ДЭК с фенолом. В реактор с неподвижным слоем загружали 8,4 г (25 мл) катализатора С. В ребойлер загружали 287 г фенола и 530 г ДЭК. Испытание проводили при следующих условиях. Давление верхней части колонны: 17-19 фунт/кв.дюйм изб. Температура ребойлера: 345-348F. Температура дистилляционной колонны: 270-285F. Скорость рециркуляции: 66-67 мл/мин. Давление наверху реактора с неподвижным слоем: 19-21 фунт/кв.дюйм изб. Давление внизу реактора с неподвижным слоем: 19-22 фунт/кв.дюйм изб. Температура реактора с неподвижным слоем: 331-335F. Скорость потока азота в ребойлер: 60 мл/мин. 0 флегмы из сборника орошающей фракции. Скорость потока PhOH/ДЭК раствора: 0,18-0,19 мл/мин. Скорость нижнего потока: 0,18-0,20 мл/мин. В процессе работы поток ДЭК падал до уровня жидкости 88% (условная шкала) для поддержания постоянного уровня жидкости. Работу выполняли непрерывным сливом реакционной смеси в ребойлер, в то же время закачивая 36,5 мас.% раствора фенола в ДЭК. Результаты 234-часового рабочего цикла собраны в табл. 2. Поток верхнего погона состоял в основном из ДЭК, этанола и небольших количеств фенола и ЭФК. Образцы были взяты из ребойлера и потока верхнего погона для анализа. Фенетол был единственным продуктом в нижнем потоке продукта из ребойлера. Результат данного цикла в табл. 2 ясно показывает более медленную дезактивацию и более высокую активность, чем у катализатора А в тесте 6 А. Результат данного цикла в табл. 2 ясно показывает более медленную дезактивацию и более высокую активность катализатора С, чем у катализатора А. Работу продолжали до 360 ч рабочего цикла. Наблюдалась продолжающаяся дезактивация катализатора. После 360 ч рабочего цикла работу реактора по синтезу ароматического карбоната прерывали,чтобы провести регенерацию катализатора. Регенерацию катализатора проводили при 340F и 230 фунт/кв.дюйм изб. деполимеризацией полимеров на катализаторе циркуляцией этанола через реактор в течение 17 ч. Основными продуктами реакции деполимеризации с этанолом были фенол, ДЭК и неидентифицированные побочные продукты. Работа была продолжена с регенерированным катализатором. Результат испытания регенерированного катализатора приведен в табл. 3. Результат в табл. 3 ясно показывает, что катализатор может быть регенерирован проведением деполимеризации полимеров, осажденных на катализаторе.(a) осуществление взаимодействия реагентов в нескольких реакционных зонах, включая первичную и вторичную реакционные зоны;(b) подвод к указанной первичной реакционной зоне диалкилкарбоната и ароматического гидроксисоединения;(c) осуществление взаимодействия реагентов в первичной реакционной зоне в двухфазной системе и реакционных условиях, способствующих образованию алкиларилкарбоната;(d) переэтерификацию данного диалкилкарбоната с ароматическим гидроксисоединением в первичной реакционной зоне в присутствии твердого катализатора, выбранного из группы, включающей от двух до четырех элементов групп IV, V и VI Периодической таблицы, нанесенных на пористый материал, который имеет поверхностные гидроксильные группы;(e) извлечение потока двухфазного продукта из первичной реакционной зоны;(f) отделение потока двухфазного продукта со стадии (е) для извлечения парообразного алкилового спирта и жидкого алкиларилкарбоната;(g) диспропорционирование алкиларилкарбоната во вторичной реакционной зоне в двухфазной системе в присутствии твердого катализатора, выбранного из группы, включающей от двух до четырех элементов групп IV, V и VI Периодической таблицы, нанесенных на пористый материал, который имеет поверхностные гидроксильные группы;(h) извлечение потока двухфазного продукта из вторичной реакционной зоны и(i) отделение потока двухфазного продукта со стадии (h) для извлечения парообразного компонента, включающего алкиларилкарбонат, и жидкого продукта, включающего диарилкарбонат. 2. Способ по п.1, в котором диалкилкарбонат представляет собой диэтилкарбонат (ДЭК), ароматическое гидроксисоединение представляет собой фенол, алкиларилкарбонат представляет собой этилфенилкарбонат (ЭФК) и диарилкарбонат представляет собой дифенилкарбонат (ДФК). 3. Способ по п.2, в котором на стадии (g) мольное отношение фенола к ДФК находится в диапазоне от 0,05 до 10. 4. Способ по п.1, в котором твердый катализатор на стадии (d) представляет собой оксиды, гидроксиды, оксигидроксиды, алкоксиды или их смеси элементов из групп IV, V и VI. 5. Способ по п.4, в котором носитель катализатора включает обработанный кремнезем. 6. Способ по п.4, в котором твердый катализатор на стадии (g) представляет собой оксиды, гидроксиды, оксигидроксиды, алкоксиды или их смеси элементов из групп IV, V и VI. 7. Способ по п.5, в котором твердый катализатор на стадии (d) выбирают из группы, состоящей из металлических алкоксидов и смешанных металлических алкоксидов групп IV и V. 8. Способ по п.4, в котором твердый катализатор на стадии (d) представляет собой TiO2/Nb2O5, нанесенный на обработанный кремнезем, причем указанный обработанный кремнезем имеет менее чем 0,05 мас.% Na. 9. Способ по п.1, в котором твердый катализатор на стадии (g) представляет собой оксиды, гидроксиды, оксигидроксиды, алкоксиды или их смеси элементов из групп IV, V и VI.- 15014058 10. Способ по п.9, в котором носитель катализатора включает обработанный кремнезем. 11. Способ по п.10, в котором твердый катализатор на стадии (g) выбирают из группы, состоящей из металлических алкоксидов и смешанных металлических алкоксидов групп IV и V. 12. Способ по п.9, в котором твердый катализатор на стадии (g) представляет собой TiO2/Nb2O5, нанесенный на обработанный кремнезем, причем указанный обработанный кремнезем имеет менее чем 0,05 мас.% Na. 13. Способ по п.1, в котором процесс на стадии (g) выполняют в присутствии ароматического гидроксисоединения при мольном отношении ароматического гидроксисоединения к ДФК в диапазоне от 0,05:1 до 10:1. 14. Способ по п.13, в котором процесс на стадии (g) выполняют в присутствии воды в количестве до 0,3 мас.%. 15. Способ по п.1, в котором процесс на стадии (d) выполняют в присутствии ароматического гидроксисоединения при мольном отношении ароматического гидроксисоединения к ДЭК выше 0,2. 16. Композиция твердого катализатора диспропорционирования или переэтерификации, выбранного из группы, состоящей из оксидов, гидроксидов, оксигидроксидов и алкоксидов от двух до четырех элементов из групп IV, V и VI Периодической таблицы, нанесенного на пористый материал, который имеет поверхностные гидроксильные группы. 17. Композиция твердого катализатора по п.16, в которой указанный носитель предварительно подвергают обработке, чтобы увеличить число содержащихся на нем гидроксильных групп. 18. Композиция твердого катализатора по п.17, в которой указанная обработка включает контактирование указанного носителя с раствором основания. 19. Композиция твердого катализатора по п.18, в которой указанный носитель включает кремнезем.
МПК / Метки
МПК: C07C 69/96, B01J 23/20, B01J 21/06
Метки: карбонатов, производства, способ, органических
Код ссылки
<a href="https://eas.patents.su/17-14058-sposob-proizvodstva-organicheskih-karbonatov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ производства органических карбонатов</a>
Применение карбонатов четвертичного аммония в качестве антикоррозионных агентов, способ ингибирования коррозии и антикоррозионные покрытия, в которых применяются эти агенты

Номер патента: 13838
Опубликовано: 30.08.2010
Авторы: Чян Майкл И., Холл Ларри К., Кимлер Джозеф, Шеблейн Джозеф У.
МПК: C09D 5/08, C23F 11/14, C23G 1/14...
Метки: способ, четвертичного, аммония, покрытия, агенты, применяются, карбонатов, антикоррозионных, антикоррозионные, ингибирования, качестве, эти, агентов, которых, коррозии, применение
Формула / Реферат:
1. Способ ингибирования коррозии металлической подложки, включающий стадию приведения подложки в соприкосновение с композицией, включающей по меньшей мере один карбонат четвертичного аммония, обладающий формулойв которой R1 обозначает C1-C20-алкильную группу и R2обозначает С1-С20-алкильную группу.2. Способ по п.1, в котором композиция дополнительно включает по меньшей мере один бикарбонат четвертичного аммония, обладающий формулойв которой R1...
Способ переработки органических отходов в моторные топлива

Номер патента: 8270
Опубликовано: 27.04.2007
Автор: Ангелов Чавдар Ангелов
МПК: C07C 1/04, C10J 3/64, C10J 3/18...
Метки: переработки, способ, топлива, моторные, органических, отходов
Формула / Реферат:
1. Способ переработки органических отходов в моторные топлива, включающий стадию обработки отходов газифицирующим агентом, содержащим кислород, водяной пар и/или диоксид углерода, в результате чего получают синтез-газ, который затем компримируют, подвергают глубокой очистке от механических примесей и соединений серы, азота и тяжелых металлов, далее очищенный синтез-газ или синтез-газ с жидкими органическими отходами подают в реактор синтеза...
Способ удаления тяжелых металлов из органических соединений

Номер патента: 2285
Опубликовано: 28.02.2002
Авторы: Аллегрини Пьетро, Вилла Марко, Канната Винченцо, Роси Алессандро
МПК: C07B 63/00
Метки: металлов, удаления, способ, органических, соединений, тяжелых
Формула / Реферат:
1. Способ удаления тяжелых металлов из органических соединений, отличающийся тем, что раствор органического соединения в растворителе, не смешивающемся с водой, обрабатывается производным цистеина формулы где R представляет атом водорода, линейную или разветвленную C1-С6 ацильную группу или бензоильную группу. 2. Способ по п.1, где молярное соотношение производного цистеина и тяжелого металла составляет от 1:1 до 100:1. 3. Способ по п.2, где...
Способ и установка для выделения органических веществ из содержащей их газовой смеси

Номер патента: 4384
Опубликовано: 29.04.2004
Авторы: Шуммер Гунтер, Штуве Арнд, Принц Петер, Шмитц Йозеф, Штробель Райнер
МПК: C07B 63/00, B01D 5/00, C07C 255/08...
Метки: смеси, газовой, веществ, содержащей, способ, установка, органических, выделения
Формула / Реферат:
1. Способ выделения одного или нескольких органических веществ из содержащей их газовой смеси, которую подвергают быстрому охлаждению в колонне, отличающийся тем, что быстрое охлаждение газовой смеси проводят в верхней части колонны, а в нижней части колонны подвергают отгонке охлаждающую жидкость смеси. 2. Способ по п.1, отличающийся тем, что быстрое охлаждение подаваемой в колонну газовой смеси проводят путем непосредственного воздействия на...
Способ и технологический блок для получения синтез-газа для дальнейшего производства аммиака

Номер патента: 777
Опубликовано: 24.04.2000
Авторы: Стал Хенрик Отто, Лаурсен Карстен Лау
Метки: блок, производства, аммиака, способ, технологический, получения, синтез-газа, дальнейшего
Формула / Реферат:
1. Способ получения синтез-газа, предназначенного для дальнейшего производства аммиака, из углеводородного сырья, предусматривающий последовательные этапы первичного и вторичного каталитического парового реформинга сырья в первичном теплообменном паровом реформинг-реакторе и во вторичном реформинг-реакторе, и в котором выходящий поток газа, подвергнутый первичному паровому реформингу, нагревают непрямым теплообменом горячим потоком газообразных...