Циклизация пептидов







Формула / Реферат
1. Способ циклизации пептида путем лактамизации, заключающийся в том, что:
а) удаляют у пептида защитные группы аллильного типа путем катализа в присутствии Pd(0) и акцептора аллила, где пептид защищен лабильной в основных условиях защитной группой на его Na и содержит, кроме того, по меньшей мере одну защищенную аллилоксикарбонилом аминофункцию боковой цепи лизина или аналога такой боковой цепи лизина и, кроме того, содержит по меньшей мере одну защищенную сложным аллиловым эфиром w-карбоксильную группу боковой цепи глутамила или аспартила или аналогов таких боковых цепей, при сохранении лабильной в основных условиях защитной группы на Na , где аллильные защитные группы являются замещенными или не имеют заместителей,
б) осуществляют циклизацию пептида путем лактамизации боковых цепей с удаленными защитными группами в присутствии реагента, представляющего собой слабое основание, и
в) удаляют у пептида лабильную в основных условиях защитную группу на его Na.
2. Способ по п.1, отличающийся тем, что Na, защищенный лабильной в основных условиях защитной группой, представляет собой Na аспартильного остатка, имеющего дополнительную защищенную аллиловым сложным эфиром карбоксильную группу, и где предпочтительно аспартильный остаток представляет собой фрагмент дипептидной последовательности Asp-His.
3. Способ по п.1 или 2, отличающийся тем, что на дополнительной конечной стадии г) осуществляют дальнейшее удлинение пептида путем продолжения синтеза пептида или сочетания пептидного фрагмента с Na с удаленной защитной группой.
4. Способ по п.3, отличающийся тем, что пептид связывают с твердой фазой в процессе циклизации при условии, что аллильные защитные группы не служат в качестве "хэндла" смолы и что удлинение пептида на стадии продолжают путем твердофазного синтеза пептида.
5. Способ по п.1 или 2, отличающийся тем, что стадию циклизации б) осуществляют в присутствии реагента, представляющего собой слабое основание, в апротонном полярном растворителе и, кроме того, в присутствии связывающего агента.
6. Способ по п.5, отличающийся тем, что реакцию циклизации осуществляют в присутствии ГХТУ или ТХТУ или фосфониевой соли бензотриазола в качестве связывающего агента.
7. Способ по п.6, отличающийся тем, что пептид связывают со смолой, представляющей собой подложку, с помощью лабильной в кислотных условиях связи.
8. Способ по одному из пп.1-5, отличающийся тем, что пептид ковалентно связывают на С-конце с помощью лабильной в кислотных условиях связи.
9. Способ по п.1, отличающийся тем, что лабильная в основных условиях защитная группа представляет собой группу FMOC.
10. Способ по п.1 или 7, отличающийся тем, что защитные аллильную и Alloc-группы удаляют с помощью катализа с использованием Pd(0) в присутствии акцептора аллила, предпочтительно с использованием избытка R1-PhnSiHm в качестве акцептора аллила, где R1 является заместителем в ароматическом ядре и представляет собой арил, алкил или аралкил, n обозначает 1 или 2 и m обозначает 2 или 3, более предпочтительно где акцептор аллила представляет собой PhSiH3.
11. Циклический пептид формулы II или III, имеющий Na, который защищен лабильной в основных условиях защитной группой


в которой Y обозначает лабильную в основных условиях защитную группу, n равно 1-10, предпочтительно n равно 1 или 2, наиболее предпочтительно n равно 1, кроме того, в которой m равно 1-15, предпочтительно m равно 3-6, наиболее предпочтительно m равно 3, кроме того, в которой х равно 1-200 и q равно 0-200, предпочтительно х равно 3-50 и q равно 0-50, где R1 и R2, каждый, обозначают встречающуюся в естественных условиях аминокислотную боковую цепь или ее не встречающееся в естественных условиях производное, где боковая цепь может содержать, кроме того, защитную группу, за исключением защитных групп, представляющих собой простой аллиловый эфир и аллилоксикарбонил, и в которой А обозначает смолу или "хэндл" смолы или в которой R2 необязательно может представлять собой встречающуюся в естественных условиях аминокислотную боковую цепь или ее не встречающееся в естественных условиях производное, где боковая цепь связана со смолой или "хэндлом" смолы через простой эфир, тиоэфир, сложный эфир, сложный тиоэфир, амидогруппу или фрагмент вторичной или третичной аминогруппы, при условии, что тогда А выбирают из ряда, включающего ОН, NH2, NR'1H или NR'1R'2, OR'3, где R'1 и R'2 независимо друг от друга обозначают С1-С4алкил.
12. Пептид по п.11, отличающийся тем, что лабильная в основных условиях группа представляет собой FMOC.
13. Пептид по п.11 или 12, отличающийся тем, что пептид имеет формулу II, в которой n обозначает 1 или 2, более предпочтительно n обозначает 1.
14. Пептид по одному из пп.11-13, отличающийся тем, что А представляет собой "хэндл" смолы или связывающий фрагмент смолы за исключением "хэндла" смолы, представляющего собой аллилоксикарбонильный фрагмент.
15. Пептид по п.14, отличающийся тем, что смола или "хэндл" смолы имеет формулу IV

в которой R'" обозначает смолу и R"1, R"2, R''3 независимо обозначают водород, С1-С4алкил или С1-С4алкоксигруппу и могут иметь одинаковые или различные значения, при условии, что только один из R"1, R"2 может обозначать водород, и где L обозначает кислород, серу, азот или имеет формулу V

16. Пептид по п.15, отличающийся тем, что смола или "хэндл" смолы имеет формулу VI, в которой радикалы R'", R"1, R''2 имеют указанные выше значения, при условии, что L выбирают из группы, включающей кислород и азот,

17. Пептид по п.16, отличающийся тем, что смола или "хэндл" смолы имеет формулу VII, в которой радикалы R'", R"1, R''2 имеют указанные выше значения, при условии, что L выбирают из группы, включающей кислород и азот,

18. Пептид по п.17, отличающийся тем, что когда L обозначает кислород, R"1, R"2 независимо обозначают водород, метил или метоксигруппу, при условии, что только один из R"1, R"2 может обозначать водород и, когда L обозначает азот, независимо обозначают метил или метоксигруппу, предпочтительно метоксигруппу.
19. Пептид по п.18, отличающийся тем, что L обозначает кислород, R"1 обозначает водород, R"2 обозначает метил или метоксигруппу и А предпочтительно обозначает смолу или "хэндл" смолы.
20. Пептид по п.19, отличающийся тем, что R"2 обозначает метил.
21. Циклический пептид формулы II или III, имеющий Na, который защищен лабильной в основных условиях защитной группой


в которой Y обозначает лабильную в основных условиях защитную группу, n равно 1-10, предпочтительно n равно 1 или 2, наиболее предпочтительно n равно 1, кроме того, в которой m равно 1-15, предпочтительно m равно 3-6, наиболее предпочтительно m равно 3, кроме того, в которой х равно 1-200 и q равно 0-200, где R1 и R2, каждый независимо, обозначают встречающуюся в естественных условиях аминокислотную боковую цепь или ее не встречающееся в естественных условшях производное, где боковая цепь может содержать, кроме того, защитную группу, за исключением защитных групп, представляющих собой простой аллиловый эфир и аллилоксикарбонил, и в которой А обозначает смолу или "хэндл" смолы или в которой R2 необязательно представляет собой встречающуюся в естественных условиях аминокислотную боковую цепь или ее не встречающееся в естественных условиях производное, где боковая цепь связана со смолой или "хэндлом" смолы через простой эфир, тиоэфир, сложный эфир, сложный тиоэфир, амидогруппу или фрагмент вторичной или третичной аминогруппы, при условии, что тогда А представляет собой OR'3, где R'3 обозначает защитную группу, за исключением аллильных групп, предпочтительно R'3 обозначает трет-бутил или пентафторфенил.
22. Применение комплексов Pd(P[орто-толил]3)2 или Pd(P[2,4-ксилоил]3)2 для катализа удаления защищенных аллилом карбоксильных групп, или защищенных аллилоксикарбонилом аминогрупп, и/или гидроксигрупп.
23. Применение по п.22, отличающееся тем, что в качестве акцептора или поглощающего агента аллильной группы используют органический сульфинат.
24. Применение по п.21 или 22, отличающееся тем, что защищенные аллилом или аллилоксикарбонилом группы представляют собой фрагмент необязательно имеющего другие защитные группы пептида, предпочтительно FMOC-защищенного пептида, где группа FMOC связана с Na пептида.
25. Способ циклизации пептида путем лактамизации, заключающийся в том, что:
а) удаляют у пептида защитные группы аллильного типа путем катализа в присутствии Pd(0) и акцептора аллила, в котором пептид защищен лабильной в основных условиях защитной группой на его Na, который представляет собой Na аспартильного остатка, имеющего защищенную сложным аллиловым эфиром w-карбоксильную группу и содержит, кроме того, по меньшей мере одну защищенную аллилоксикарбонилом аминофункцию боковой цепи лизина или аналога такой боковой цепи лизина при сохранении лабильной в основных условиях защитной группы на Na, где аллильные защитные группы являются замещенными или не имеют заместителей,
б) осуществляют циклизацию пептида путем лактамизации боковых цепей с удаленными защитными группами в присутствии реагента, представляющего собой слабое основание, и
в) удаляют у пептида лабильную в основных условиях защитную группу на его Na.
Текст
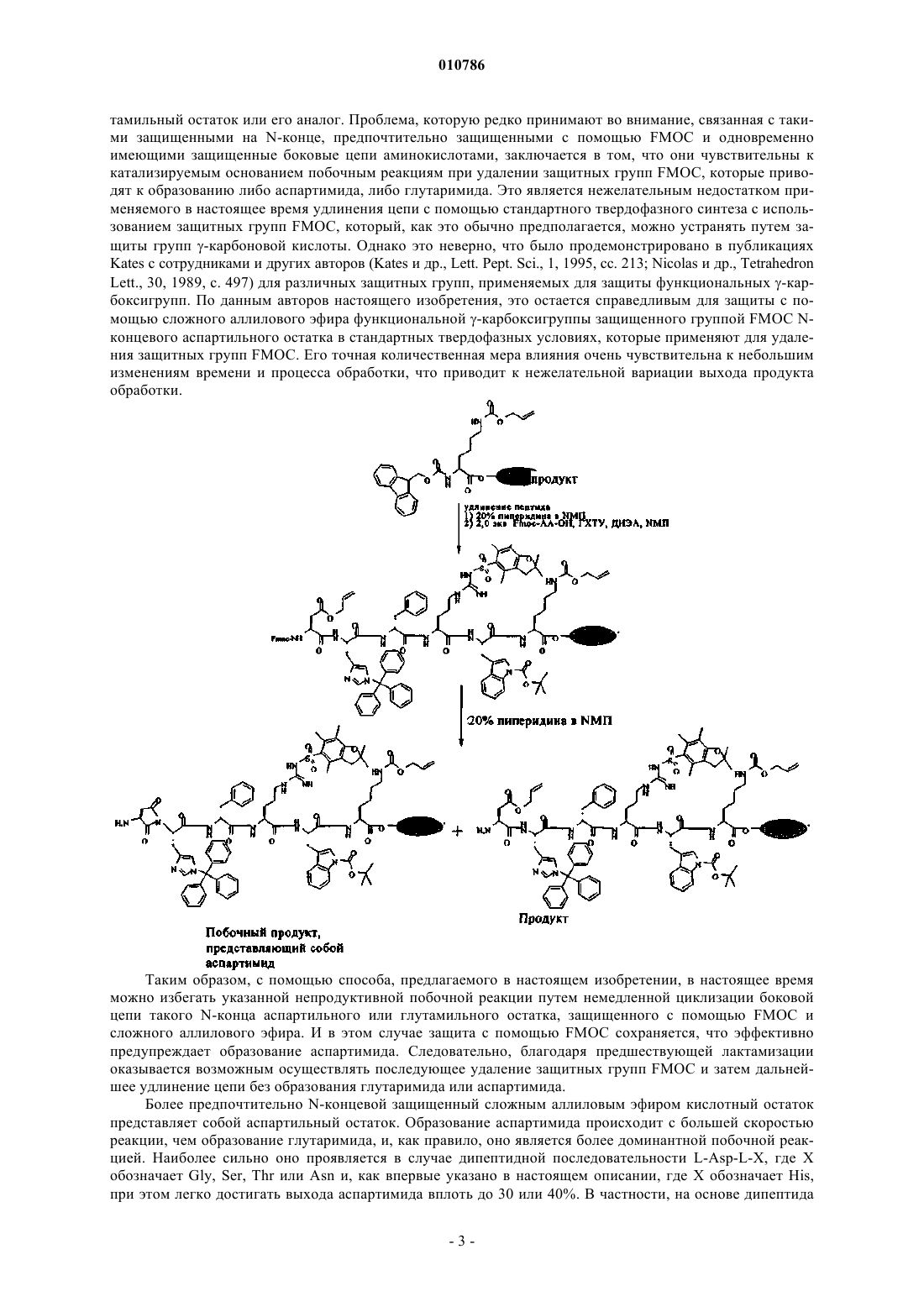
010786 Настоящее изобретение относится к способу синтеза циклического пептида, а именно циклического пептида, содержащего кольцо, замкнутое через карбоксильную группу боковой цепи одного аминокислотного остатка и аминогруппу боковой цепи второго аминокислотного остатка. В научной литературе описана циклизация пептида на смоле путем образования лактама через карбоксильную группу аминокислотной боковой цепи (т.е. карбоксильную группу боковой цепи вне зависимости от длины углеродной цепи), как правило, аспартильного или глутамильного остатка, и -аминогруппу аминокислотной боковой цепи (т.е. аминогруппу боковой цепи вне зависимости от длины углеродной цепи), как правило, остатка лизина. У Rijkers и др., An optimized solid phase strategy - including on-resin lactamization - of Astressin, itsretro-, inverso- and retro-inverso isomers, Biopolymers 63, 2002, cc. 141-149, описана лактамизация Вос-защищенного 41-мерного пептида, связанного с амидной смолой Ринка. Для этого осуществляли удаление защитных групп в положениях 30 (Glu) и 33 (Lys) путем одностадийного катализируемого Pd(0) удаления защитных аллилльных и Alloc-групп и затем циклизацию в присутствии BOP/HOBt и основания Хюнига в N-метилпирролидоне. Пептид синтезировали согласно стандартной ортогональной схеме защиты,основанной на применении FMOC для всех остатков, за исключением последнего, который защищали с помощью Boc. Затем N-концевую защитную группу Boc удаляли путем кислотной обработки ТФК, одновременно высвобождая пептид из смолы. Недостаток этого метода заключается в том, что предпосылкой для последующей циклизации сегмента пептида является прекращение синтеза полноразмерного пептида. Таким образом, происходит потеря более ценного полноразмерного пептида вследствие нежелательных побочных реакций (образование пироглутамата) или неполного удаления защитных аллильных/Alloc-групп. Кроме того, не всегда целесообразно применять лабильную в кислых условиях защитную группу при использовании смолы,поскольку это препятствует последующей дополнительной дериватизации пептида на смоле, напримерN-концевому блокированию путем ацетилирования. До N-концевого ацетилирования нельзя осуществлять никаких реакций, поскольку это делает концевой N чувствительным к эпимеризации на хиральном С. У Kates и др., A novel, convenient three dimensional orthogonal strategy for solid-phase synthesis of cyclicpeptides, Tetrahedron Letters 34, 1993, cc. 1549-1552, описана циклизация типа голова к хвосту декамерного пептида путем присоединения (заякоривания) С-концевого аспартильного или глутамильного остатка к различным элементам смолы, обеспечивающим прикрепление пептида (хэндлам смолы (resinhandles, где остаток защищен на его С-конце защитной группой аллилового сложного эфира. После линейного твердофазного синтеза полной пептидной цепи с использованием защитной группы FMOC фрагмент сложного аллилового эфира удаляют путем катализа в присутствии Pd, после чего последовательно удаляют N-FMOC и затем осуществляют опосредуемую BOP/HOBt/DIEA циклизацию типа голова к хвосту. Интересно отметить, что было установлено, что стратегия, в которой С-концевой аспартильный остаток с удаленной защитной группой (Asn8, образованный в результате сочетания FMOC-Asp с хэндлом смолы PAL) подвергали сочетанию с N N-концевого аспартильного остатка с удаленной защитной группой в отсутствие защитной группы FMOC, оказалась более эффективной по сравнению с другими стратегиями синтеза, основанными на пермутации исходной точки синтеза вдоль последовательности циклического пептида. Недостатком, ограничивающим применение этого метода, является то, что в нем по умолчанию подразумевается, что в ходе основной стадии удаления аллильной защитной группы происходит в качестве побочной реакции частичное удаление FMOC, обусловленное присутствием нуклеофильных реагентов, и поэтому оно совершенно не оказывает влияния на реакционную схему. Затем так или иначе последовательно происходит дальнейшее завершение удаления защитных групп FMOC, и оно требуется для осуществления последующего связывания пептида по типу голова к хвосту. В отличие от этого, циклизация только путем лактамизации функциональных групп пептидной боковой цепи принципиально зависит от сохранения полной защиты N. У Blankemeyer и др., Tetrahedron Lett. 29, 1988, сс. 5871-5874, описан синтез нескольких защищенных пептидных фрагментов на целлюлозных дисках, содержащих 4-(4'-метокситритилокси)бут-2-енилоксигексановую кислоту в качестве аллильного хэндла к твердой фазе, в котором, кроме того, используется химия с применением FMOC. Отщепление линкера, представляющего собой сложный аллиловый эфир, осуществляли путем обработки Pd(PPh3)4 в безводном ТГФ и последующего добавления раствораHOBt в ТГФ. При создании настоящего изобретения был разработан другой способ циклизации пептидной боковой цепи, лишенный недостатков, присущих известным из существующего уровня техники методам, который особенно пригоден для циклизации пептидов, содержащих аспартильные боковые цепи. Таким образом, в настоящем изобретении предложен способ циклизации пептида, заключающийся в том, что: а) удаляют у пептида защитные группы аллильного типа, где пептид защищен лабильной в основных условиях защитной группой на его N и где пептид содержит, кроме того, по меньшей мере одну защищенную аллилоксикарбонилом аминофункцию боковой цепи лизина (т.е. -аминогруппу) или ана-1 010786 лога такой боковой цепи лизина и, кроме того, содержит по меньшей мере одну защищенную сложным аллиловым эфиром -карбоксильную группу боковой цепи глутамила (т.е. -карбоксигруппу), или аспартила (т.е. -карбоксигруппу), или аналогов таких боковых цепей, при сохранении лабильной в основных условиях защитной группы на N,б) осуществляют циклизацию пептида путем лактамизации боковых цепей с удаленными защитными группами в присутствии реагента, представляющего собой слабое основание, и в) удаляют у пептида лабильную в основных условиях защитную группу на его N,при дополнительном условии, что аллильные защитные группы могут не иметь заместителей или могут быть дополнительно замещены алкилом или аралкилом, которые, в свою очередь, могут не иметь заместителей или могут быть дополнительно замещены галогеном или алкоксигруппой. В предпочтительном обычном варианте осуществления изобретения для защиты -аминофункции применяют стандартную незамещенную защитную группу Alloc (т.е. аллилоксикарбонил или проп-2-енилоксикарбонил),а -карбоксигруппу защищают путем этерификации с образованием сложного эфира с использованием аллилокси(пропен-2-окси)группы. В контексте настоящего описания понятие -карбоксильная группа аминокислотной боковой цепи обозначает концевую карбоксильную группу боковой цепи вне зависимости от длины углеродной цепи, а понятие -аминогруппа обозначает концевую аминогруппу боковой цепи вне зависимости от длины углеродной цепи. Их аналогами могут быть, например, их изомеры боковой цепи. Например,пригодными аналогами встречающегося в естественных условиях лизина являются - или -аминоизомер лизина. В контексте настоящего описания понятие боковая цепь применительно к аминокислоте или производному аминокислоты используется согласно соответствующему определению ИЮПАК-ИЮБ(Международный союз по теоретической и прикладной химии и Международный биохимический союз/Объединенная комиссия по биохимической номенклатуре (International Union of Pure and AppliedChemistry and International Union of Biochemistry/Joint Commission on Biochemical Nomenclature),Nomenclature and Symbolism for Amino Acids and Peptides, Pure Appl. Chem, 56, 1984, cc. 595-624). Эта реакционная схема не была описана ранее. При создании изобретения неожиданно было установлено, что после удаления защитных аллильных групп целостность группы FMOC в значительной степени сохраняется, в частности, когда удаление защитных аллильных групп осуществляют у пептида, связанного с твердой фазой, и вследствие этого циклизацию также осуществляют на материале, связанном с твердой фазой. Предпочтительно после завершения последовательности реакций удлинение пептидной цепи продолжают, более предпочтительно продолжают на твердой фазе, так как при этом имеется преимущество, заключающееся в том, что после предшествующей стадии циклизации не требуется последующего применения условий ограничивающих разведений, которые благоприятствуют осуществлению внутримолекулярной циклизации по сравнению с межмолекулярной циклизацией. Поэтому предлагаемая в настоящем изобретении реакция должна быть наиболее целесообразной в том случае, когда в конечной пептидной цепи присутствуют многочисленные глутамильные и/или аспартильные остатки и остатки лизина или их аналоги, такие, например, как нор- или гомолизин, хотя для циклизации первого синтезированного фрагмента конечной полноразмерной пептидной цепи планируется только специфическое спаривание этих остатков. Такой подход, основанный на местной лактамизации, должен быть наиболее целесообразным, когда полноразмерный пептид содержит несколько последующих субдоменов или пептидных петлеобразных структур в случае биологически активых пептидов, которые нужно стабилизировать с помощью циклизации боковой цепи. Лактамизация представляет собой более стабильный менее чувствительный к окислительно-восстановительным процессам вариант по сравнению с встречающимися в естественных условиях стабилизированными путем образования дисульфидных мостиков вариантами или не встречающимися в естественных условиях химическими аналогами на основе аналогов аминокислот. Последние из указанных функциональных групп, как правило, оказываются более иммуногенными in vivo, в то время как согласно настоящему изобретению можно применять аналоги встречающихся в естественных условиях аминокислот в качестве более совершенного варианта. В предпочтительном варианте осуществления изобретения пептид содержит только один защищенный аллилом остаток лизина и только один защищенный аллилом аспартильный или глутамильный остаток и, следовательно, существует один и только один вариант связывания путем циклизации, позволяющий получать гомогенный продукт. В отличие от методов, известных из существующего уровня техники, пептид, получаемый согласно способу, предлагаемому в настоящем изобретении, можно применять для дальнейшего N-концевого удлинения пептида, необязательно и предпочтительно на твердой фазе, осуществляя это путем последовательного ступенчатого добавления или модификации аминокислотных остатков или других групп, например путем добавления новых N-защищенных аминокислот или с помощью реакции конденсации Nконца с другим пептидом. В другом особенно предпочтительном варианте осуществления изобретения N-концевой остаток защищенного пептида, имеющий лабильную в основных условиях защитную группу на его N, представляет собой по меньшей мере один защищенный сложным аллиловым эфиром аспартильный или глу-2 010786 тамильный остаток или его аналог. Проблема, которую редко принимают во внимание, связанная с такими защищенными на N-конце, предпочтительно защищенными с помощью FMOC и одновременно имеющими защищенные боковые цепи аминокислотами, заключается в том, что они чувствительны к катализируемым основанием побочным реакциям при удалении защитных групп FMOC, которые приводят к образованию либо аспартимида, либо глутаримида. Это является нежелательным недостатком применяемого в настоящее время удлинения цепи с помощью стандартного твердофазного синтеза с использованием защитных групп FMOC, который, как это обычно предполагается, можно устранять путем защиты групп -карбоновой кислоты. Однако это неверно, что было продемонстрировано в публикацияхLett., 30, 1989, с. 497) для различных защитных групп, применяемых для защиты функциональных -карбоксигрупп. По данным авторов настоящего изобретения, это остается справедливым для защиты с помощью сложного аллилового эфира функциональной -карбоксигруппы защищенного группой FMOC Nконцевого аспартильного остатка в стандартных твердофазных условиях, которые применяют для удаления защитных групп FMOC. Его точная количественная мера влияния очень чувствительна к небольшим изменениям времени и процесса обработки, что приводит к нежелательной вариации выхода продукта обработки. Таким образом, с помощью способа, предлагаемого в настоящем изобретении, в настоящее время можно избегать указанной непродуктивной побочной реакции путем немедленной циклизации боковой цепи такого N-конца аспартильного или глутамильного остатка, защищенного с помощью FMOC и сложного аллилового эфира. И в этом случае защита с помощью FMOC сохраняется, что эффективно предупреждает образование аспартимида. Следовательно, благодаря предшествующей лактамизации оказывается возможным осуществлять последующее удаление защитных групп FMOC и затем дальнейшее удлинение цепи без образования глутаримида или аспартимида. Более предпочтительно N-концевой защищенный сложным аллиловым эфиром кислотный остаток представляет собой аспартильный остаток. Образование аспартимида происходит с большей скоростью реакции, чем образование глутаримида, и, как правило, оно является более доминантной побочной реакцией. Наиболее сильно оно проявляется в случае дипептидной последовательности L-Asp-L-X, где X обозначает Gly, Ser, Thr или Asn и, как впервые указано в настоящем описании, где X обозначает His,при этом легко достигать выхода аспартимида вплоть до 30 или 40%. В частности, на основе дипептида-Asp-Gly- очень легко может образовываться аспартимид, что, как правило, требует применения дополнительной защитной группы HMB на остатке глицина, создающей препятствие для образования аспартимида, как это описано у Packman и др., Tetrahedron Lett., 36, 1995, с. 7523. Однако использование дипептидов, имеющих Gly(HMB), является чрезвычайно дорогим, и поэтому желательно его избегать. Таким образом, в этих случаях настоящее изобретение даже обладает дополнительным преимуществом по сравнению с существующим уровнем техники. Стадию удаления защитных аллильных и/или Alloc-групп (в дальнейшем для краткости называемую удалением аллила) можно осуществлять известными в данной области методами, такими, например,как гидростаннолиз с помощью Bu3SnH или обработка тетракис(трифенилфосфин)палладием(0)[Pd(PPh3)4] в ТГФ в практически нейтральных условиях в присутствии нуклеофила, такого, например,как морфолин, димедон, N-метилаланин, HOBt, боргидрид или N,N'-диметилбарбитуровая кислота, в качестве аллильного акцептора. Существуют варианты этих методов, в которых используют различные значения рН, но, конечно, при этом необходимо соблюдать осторожность, учитывая чувствительность связи или хэндл-групп смолы к значению рН и чувствительность защитной группы к действию основания. Удаление защитных аллильных групп предпочтительно проводят путем катализа в присутствии реактивных в отношении аллильной группы палладиевых комплексов, предпочтительно комплексов на основе Pd(0), предпочтительно палладиевых комплексов, имеющих C1-С 10 триалкилфосфитные, С 3-С 10 трициклоалкилфосфитные, или триарилфосфиновые, или тригетероарилфосфиновые лиганды, где арил или гетероарил может быть дополнительно замещен электронодонорными заместителями или не иметь их, более предпочтительно палладиевых комплексов, имеющих фенилфосфиновые лиганды, где фенил может быть дополнительно замещен С 1-С 5 алкилом, где предпочтительно фенил представляет собой толил или ксилоил, более предпочтительно представляет собой фенил, 2,4-ксилоил или ортотолил. Предпочтительно фосфиновые лиганды представляют собой монофосфиновые лиганды, более предпочтительно нехелатирующие одновалентные лиганды. Хотя палладиевые комплексы предпочтительно представляют собой монопалладиевые комплексы, следует понимать, что понятие комплекс относится также к дипалладиевым и высшим палладиевым комплексам, хотя более предпочтительными являются монопалладиевые комплексы. Согласно настоящему изобретению наиболее предпочтительным является применение тетракис(трифенилфосфин)палладия, [Pd(PPh3)4], в присутствии акцептора, или поглотителя аллила, или соответствующих комплексов Pd(P[ортотолил]3)2 или Pd(Р[2,4-ксилоил]3)2, или смешанных комплексов, которые могут иметь как трифенилфосфиновые, так и три(ортотолил)фосфиновые лиганды. По сравнению с трифенилфосфиновыми метилфенилфосфиновые лиганды и, прежде всего, триортотолилфосфиновые лиганды улучшают скорость каталитической реакции, кроме того, позволяя уменьшать общее количество прецизионного металлического катализатора, применяемого для поддержания оптимальных выходов. Описание катализируемого Pd(0) удаления защитных аллильных и аллилоксигрупп приведено у Jeffrey и др., J. Org. Chem., 47, 1982, cc. 587-590. Как указано у Jeffrey, можно, и это является общепринятым в данной области, создавать комплексы, предлагаемые в настоящем изобретении, в виде обменного катализатора in situ, а именно путем смешения менее стабильно координированных Pd-комплексов с предпочтительными лигандами, предлагаемыми в настоящем изобретении. Следовательно,примерами пригодных каталитических комплексов, отличных от наиболее предпочтительного Pd(PPh3)4,являются PdCl2(PPh3)2/PPh3, PdCl2(PPh3)2/P(oTol)3, Pd(DBA)2/P(oTol)3 или Pd[P(oTol)3]2 (Organometallics,14 (6), 1995, cc. 3030-3039), Pd(OAc)2/триэтилфосфит, Pd(OAc)2/PPh3 или Pd(OAc)2/P(oTol)3. Предпочтительно первоначально добавляемые комплексы на основе Pd(0) (Pd(0)-комплексы) позволяют создавать каталитические комплексы in situ благодаря присутствию пригодных свободных лигандов, таких какPPhh3 или P(oTol)3 [oTol=ортотолил]. В целом, не является обязательным добавлять свободный лиганд для обмена с катализатором, хотя, прежде всего, при использовании с самого начала обладающих высокой активностью акриловых фосфинпалладиевых комплексов этот вариант заслуживает внимания в случае Pd(PPh3)4 с нулевой валентностью, который впервые применяли Jeffrey и др. (выше) с добавлением дополнительной порции PPh3 в реакционную смесь. Общий механизм катализируемого Pd удаления защитных групп в присутствии аллильного акцептора, как это хорошо известно в данной области, заключается в реакции переноса ацильной группы (трансацилирование). Поэтому выбор реагента, представляющего собой акцептор или поглотитель аллила, также важен для осуществления количественного удаления защитных групп в мягких реакционных условиях с избеганием при этом нежелательных побочных реакций. Пригодным поглотителем является любой нуклеофил, такой, например, как морфолин, димедон, N,N-диметилбарбитуровая кислота, метиланилин или тиосалициловая кислота.Pd(0)-Комплексы предпочтительно применяют в пригодных каталитических количествах, которые составляют от 0,005 до 0,5 экв. катализатора в пересчете на количество выделенного вещества, более предпочтительно от 0,01 вплоть до 0,1 экв. катализатора, наиболее предпочтительно от 0,015 вплоть до 0,07 экв. катализатора. Предпочтительно температура реакции составляет от 10 до 60 С, более предпочтительно от 30 до 50 С, наиболее предпочтительно примерно 40 С. В одном из предпочтительных вариантов осуществления настоящего изобретения амин-борановые комплексы применяют по меньшей мере в 1,5-2-кратном избытке по отношению к аллильной функции-4 010786 выделенного вещества в качестве нуклеофильного поглотителя аллильной группы, как это описано уcomplexes as allyl group scavengers, J. Chem. Soc., Perkin Trans. 1, 1999, cc. 2871-2874. В зависимости от точного состава аминового фрагмента, входящего в комплекс, можно обеспечивать высокие скорости превращения наряду с очень коротким временем реакции, например, с использованием трет-Bu-NH2BH3,Me2NHBH3 или NH3BH3. Из применяемого в настоящем описании определения пригодных комплексов исключены четвертичные амины, поскольку предпочтительно амин должен представлять собой первичный или вторичный алкиламин или может представлять собой аммиак. Согласно необязательному еще более предпочтительному варианту осуществления изобретения катализируемое Pd(0) удаление аллильной защитной группы осуществляют путем гидросилилолиза в присутствии донора гидрида фенилтригидросилана PhSiH3 или его функциональных производных в апротонном полярном органическом растворителе, таком, например, как дихлорметан, как это описано, в целом, у Dessolin и др., New allyl groupacceptors for palladium catalyzed transacylation of allyl carbamates, Tetrahedron Lett., 36, 1995, cc. 5741-5744. Предпочтительно в качестве акцептора аллила применяют фенилгидросилановый реагент общей формулы R1-PhnSiHm в избытке, составляющем по меньшей мере 1,5-2 экв., где R1 обозначает заместитель в ароматическом ядре и представляет собой арил, алкил или аралкил, n обозначает 1 или 2 и m обозначает 2 или 3, где акцептор аллила наиболее предпочтительно представляет собой PhSiH3. Использование как фенилсиланов, так и пригодных амин-борановых комплексов, позволяет осуществлять быстрое полное удаление защитных групп в течение промежутка времени 1 ч, как правило, в течение примерно 20-40 мин. Следовательно, они позволяют использовать мягкие условия реакции и проводить ее в течение короткого промежутка времени. Другие поглотители аллила могут требовать существенно большей продолжительности реакции. Хотя можно создавать идеальные в техническом отношении реагенты, следует отметить, что оба класса указанных выше аллильных поглотителей представляют собой ядовитые реагенты, которые могут вызывать беспокойство с точки зрения безопасности окружающей среды и безопасности при работе. В другом наиболее предпочтительном варианте осуществления изобретения, который является особенно привлекательным для промышленного производства, в качестве реагента, представляющего собой акцептор аллильной группы, применяют органический сульфинат (Honda и др., Deprotection of allyl groupswith sulfinic acids and palladium catalyst, J. Org. Chem., 62, 1997, cc. 8932-8936). При создании настоящего изобретения неожиданно было установлено, что среди реакций, которые можно использовать согласно настоящему изобретению, данная реакция позволяет получать особенно высокие выходы продукта, что,по-видимому, обусловлено стабильностью аддуктов, образующихся при этом, и мягкими реакционными условиями. Примерами таких реагентов являются 4-хлор-3-нитробензолсульфинат, 2-тиофенсульфинат,бензолсульфинат, паратолилсульфинат или гидроксиметансульфинат (также известный в форме его натриевой или цинковой соли под названием Rongalit, традиционное тривиальное название таких солей формальдегидсульфоксилаты). Органический сульфинат Ri-SO2- (или Ri-S(O)O-, соответственно) может содержать органический фрагмент любого типа, имеющий дополнительные заместители, алкил, циклоалкил, арил, гетероарил или аралкил (см. Honda и др., выше). Его можно добавлять в формах соли или кислоты. Указанный сульфинат может содержать даже конкурирующие нуклеофильные фрагменты, хотя это менее предпочтительно. Предпочтительно для достижения оптимальных выходов радикал Ri представляет собой необязательно дополнительно замещенный фенильный радикал, более предпочтительно фенильный радикал, замещенный одним или несколькими заместителями, выбранными из ряда, включающего алкил, алкилоксиалкил или алкилоксигруппу, или наиболее предпочтительно представляет собой фенил, ксилоил или толил, прежде всего, паратолил. Использование сульфинатов позволяет применять, в дополнение к более предпочтительным триарилфосфиновым комплексам, также лиганды, представляющие собой триалкил- или циклоалкилфосфитные комплексы, обеспечивающие хорошие выходы. Реакцию лактамизации осуществляют практически аналогично стандартным реакциям удлинения пептидной цепи в присутствии лабильных в основных условиях аминозащитных групп, таких как FMOC,за исключением того, что лабильная в основных условиях группа продолжает находиться на этом же пептиде, а не осуществляют добавление другой защищенной на N-конце аминокислоты, как это делают в случае удлинения цепи. Конечно, впоследствии такие же химические реакции можно применять для дальнейшего удлинения цепи на необязательной стадии г) после циклизации и удаления защитных групп. Как лактамизация, так и удлинение цепи требуют присутствия связывающего агента и, в конце концов, связывающей добавки, в зависимости от типа первичного связывающего агента или вспомогательного вещества. Связывающие агенты, применяемые для синтеза пептидов, хорошо известны в данной области (см.Bodansky M., Principles of Peptide Synthesis, 2-е изд., изд-во Springer Verlag Berlin/Heidelberg, 1993; см. также приведенное там же обсуждение роли связывающих добавок или вспомогательных веществ). Связывающие агенты могут представлять собой смешанные ангидриды (например, T3P: ангидрид пропанфосфоновой кислоты) или другие ацилирующие агенты, такие как активированные сложные эфиры или галоидангидриды (например, IBCF, изобутилхлорформиат), или они могут представлять собой карбоди-5 010786 имиды (например, 1-этил-3-(3-диметиламинопропил)карбодиимид), активированные производные бензотриазина (DEPBT: 3-(диэтоксифосфорилокси)-1,2,3-бензотриазин-4(3 Н)-он) или производные урониевой или фосфониевой соли бензотриазола. С точки зрения наиболее высокого выхода, короткого времени реакции и защиты от рацемизации в процессе удлинения цепи, наиболее предпочтительно выбирать связывающий агент из группы, включающей урониевые соли или фосфониевые соли бензотриазола, обладающие способностью активировать свободную функцию карбоновой кислоты, когда реакцию проводят в присутствии основания. Пригодными и также предпочтительными примерами таких урониевых или фосфониевых связывающих солей являются, например, HBTU (гексафторфосфат О-1 Н-бнзотриазол-1-ил)-N,N,N',N'-тетраметилурония),BOP (гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)фосфония), PyBOP (гексафторфосфат бензотриазол-1-илокситрипирролидинфосфония), PyAOP, HCTU (гексафторфосфат О-(1 Н-6-хлорбензотриазол-1-ил)-1,1,3,3-тетраметилурония), TCTU (тетрафторборат О-1 Н-6-хлорбензотриазол-1-ил)-1,1,3,3 тетраметилурония), HATU (гексафторфосфат О-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония),TATU (тетрафторборат О-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония), TOTU (тетрафторборат О[циан(этоксикарбонил)метиленамино]-N,N,N',N-тетраметилурония), HAPyU (гексафторфосфат О-(бензотриазол-1-ил)окси-бис(пирролидин)урония. Предпочтительно основный реагент представляет собой слабое основание, сопряженная с которым кислота имеет показатель кислотности pKa от 7,5 до 15, более предпочтительно от 7,5 до 10, за исключением -аминофункции пептида или аминокислоты или производного аминокислоты, и указанное основание предпочтительно представляет собой третичный пространственно затрудненный амин. Примерами такого и других предпочтительных оснований являются основание Хюнига (N,N-диизопропилэтиламин),N,N'-диалкиланилин, 2,4,6-триалкилпиридин, 2,6-триалкилпиридин или N-алкилморфолин, где алкил представляет собой С 1-С 4 алкил с прямой или разветвленной цепью, более предпочтительно он представляет собой N-метилморфолин или коллидин (2,4,6-триметилпиридин), наиболее предпочтительно он представляет собой коллидин. Применение связывающих добавок, прежде всего, связывающих добавок бензотриазольного типа,также является известным (см. Bodansky, выше). Их применение наиболее предпочтительно в том случае, когда используют связывающие агенты, представляющие собой обладающие высокой активирующей способностью урониевые или фосфониевые соли. Поэтому, кроме того, предпочтительно, чтобы добавка, служащая в качестве связывающего агента, представляла собой нуклеофильное гидроксисоединение, обладающее способностью образовывать активированные сложные эфиры, более предпочтительно имеющее кислотную нуклеофильную N-гидроксифункцию, где N обозначает имид или замещеннуюN-ацилом или N-арилом триазеногруппу, наиболее предпочтительно связывающая добавка представляет собой производное N-гидроксибензотриазола (или производное 1-гидроксибензотриазола) или производное N-гидроксибензотриазола. Такие связывающие добавки, представляющие собой N-гидроксисоединения, описаны в WO 94/07910 и ЕР-410182, соответствующее содержание которых включено в настоящее описание в качестве ссылки. Примерами таких соединений являются, например, N-гидроксисукцинимид, N-гидрокси-3,4-дигидро-4-оксо-1,2,3-бензотриазин (HOOBt), 1-гидрокси-7-азабензотриазол(HOAt) и N-гидроксибензотриазол (HOBt). Особенно предпочтительными являются производные N-гидроксибензотриазина, в наиболее предпочтительном варианте осуществления изобретения добавка, служащая в качестве связывающего агента, представляет собой гидрокси-3,4-дигидро-4-оксо-1,2,3-бензотриазин. Известны связывающие добавки, представляющие собой аммониевые соли, и их применение описано, например, в US 4806641. В следующем наиболее предпочтительном варианте осуществления изобретения связывающий агент в виде урониевой или фосфониевой соли представляет собой агент в виде урониевой соли и предпочтительно представляет собой HCTU, TCTU или HBTU, и еще более предпочтительно его применяют в сочетании с N-гидрокси-3,4-дигидро-4-оксо-1,2,3-бензотриазином или его солью. Этот вариант осуществления изобретения, в основном, предпочтительно применяют на стадии удлинения цепи пептидного синтеза после удаления лабильной в основных условиях Na-защитной группы, но его можно применять также для реакции лактамизации в процессе циклизации боковой цепи. Следует отметить, что в контексте настоящего изобретения HCTU и TCTU определены как соединения, подпадающие под понятие реагент, представляющий собой урониевую соль, несмотря на то,что с помощью анализа кристаллической структуры можно установить, что эти соединения, а также их возможные аналоги содержат изонитрозный фрагмент, а не урониевый фрагмент (О. Marder, Y. Shvo иF. Albericio HCTU and TCTU: New Coupling Reagents: Development and Industrial Applications, постер,презентация на Гордоновской конференции, февраль 2002 г.), в результате чего заместитель, представляющий собой N-амидиногруппу, приводит к образованию гуанидиниевой структуры. В контексте настоящего изобретения такой класс соединений назван подклассом гуанидиниевого типа реагентов,представляющих собой урониевые соли, предлагаемые в настоящем изобретении. Еще в одном особенно предпочтительном варианте осуществления изобретения, который, в основном, применяют для реакции лактамизации, связывающий агент представляет собой фосфониевую соль бензотриазола, такую, например, как BOP, PyBOP или PyAOP.-6 010786 Удаление защитной группы у лабильного в основных условиях N можно осуществлять стандартным в данной области методом, например с помощью 20% пиперидина в N-метилморфолине. Еще одним объектом настоящего изобретения является циклический пептид формулы II или III,имеющий N, защищенный лабильной в основных условиях защитной группой, в которой Y обозначает лабильную в основных условиях защитную группу, n равно 1-10, предпочтительно n равно 1 или 2, наиболее предпочтительно n равно 1, кроме того, в которой m равно 1-15, предпочтительно m равно 3-6, наиболее предпочтительно m равно 3, кроме того, в которой х равно 1-200 и q равно 0-200, где R1 и R2, каждый независимо, обозначают встречающуюся в естественных условиях аминокислотную боковую цепь или ее не встречающееся в естественных условиях производное, где боковая цепь может содержать, кроме того, защитную группу, за исключением защитных групп, представляющих собой простой аллиловый эфир и аллилоксикарбонил, и в которой А обозначает смолу или хэндл смолы или в которой R2 необязательно может представлять собой также встречающуюся в естественных условиях аминокислотную боковую цепь или ее не встречающееся в естественных условиях производное, где боковая цепь связана со смолой или хэндлом смолы через простой эфир, тиоэфир, сложный эфир, амидогруппу или фрагмент вторичной или третичной аминогруппы, при условии, что тогда А выбирают из ряда, включающего ОН, NH2, NR'1H или NR'1R'2, OR'3, где R'1 и R'2 независимо друг от друга обозначают С 1-С 4 алкил и R'3 обозначает защитную группу, отличную от аллильных групп, или за их исключением, предпочтительно R'3 обозначает трет-бутил или пентафторфенил. Не следует понимать, что группа боковой цепи, такая, например, как R1(x=1), обозначает один тип необязательно замещенной аминокислотной боковой цепи; это означает, что каждый остаток R1(1), R1(2) и т.д. может иметь индивидуальное значение или может иметь такое же значение, что и по меньшей мере один из других остатков. То же самое относится, конечно, и к радикалам R2(q=l), R2(q=2) и т.д. Смола или композитный элемент хэндла смолы, в принципе, может представлять собой любую смолу, применяемую для синтеза, такую, например, как полистиролдивинилбензольная смола, которую применял Мэррифилд в сочетании с гидроксибензилфенильными интегральными линкерными фрагментами, или Ванг в сочетании с гидроксибензилпарабензилоксифрагментами, такими, например, как фрагменты, к которым затем можно, например, прививать дополнительные более лабильные в основной среде линкеры, или в альтернативном варианте последние линкеры можно интегрально или непосредственно связывать со смолой. В принципе, применяемая для твердофазного синтеза смола обязательно содержит,по меньшей мере, интегральный линкер или хэндл, который является частью материала ядра твердой фазы; такой линкер или хэндл можно рассматривать как иммобилизованную защитную группуBiosciences, Германия) на основе тентагеля, которые поступают в продажу с различными привитыми хэндлами, такими как 2'-хлортритил, или смолы, которые получают путем прививки функциональными хендлами материала матрицы, такого как силикагели. Предпочтительно, когда смола является тритиловой смолой или несущей хэндл смолой, такая смола представляет собой 4-метокси- или 4,4'-диметокситритиловую смолу. Смолы, применяемые согласно настоящему изобретению, характеризуются стандартным размером ячейки сита, который составляет примерно 50-500 меш, более предпочтительно-7 010786 100-400 меш. Подразумевается, что смола или твердая фаза R'", как следует из формулы IV, содержит поперечно-сшитый полимерный материал матрицы, который может быть связан с хэндл-фрагментом,указанным в формулах IV-VII, с помощью любого химически инертного спейсера или линкера, представляющего собой алкил, алкилоксигруппу, арилоксигруппу или сложный алкиловый эфир, который следует рассматривать как интегральную часть R'". Необходимо отметить, однако, что, не говоря уже о влиянии на условия отщепления от смолы, химическая природа материала смолы и, прежде всего, химическая природа хэндл-группы может оказывать сильное влияние на эффективность сочетания при синтезе и, прежде всего, на реакции лактамизации, которое еще недостаточно изучено. Выходы зрелого пептида на стадии присутствия на смоле могут варьироваться в зависимости от типа применяемой смолы или хэндла. Поэтому в предпочтительном варианте осуществления настоящего изобретения смола или хэндл смолы имеют формулу IV, как это подробно представлено ниже в формуле изобретения, более предпочтительно формулу VI и наиболее предпочтительно формулу VII, как это подробно представлено ниже в формуле изобретения. Примерами таких смол или хэндлов смолы являются (4-метоксифенил) метил- и (4-метилфенил)метилполистирол (Atkinson и др., J. Org. Chem. 65, 2000, с. 5048), смолы, которые могут связываться с пептидным фрагментом с помощью О- или N-связи, и их производные, представляющие собой модифицированные с помощью ПЭГ смолы, соответственно. Другими примерами являются, например, лабильная в кислотных условиях смола HMPB-MBHA или HMPB-BHA (Sieber и др., Tetrahedron Lett. 28, 1987, с. 6147), лабильная в кислотных условиях амидная смола Ринка или кислотная смола Ринка (Rink и др., Tetrahedron Lett. 28, 1987, с. 3787). Понятие лабильная в кислотных условиях означает, что в течение по меньшей мере 1 ч происходит практически количественное расщепление в 2-10% ТФК в дихлорметане при температуре окружающей среды. При создании изобретения неожиданно было установлено, что использование таких предпочтительных смол, имеющих дифенилметильный структурный мотив ядра, позволяет более эффективно осуществлять реакцию сочетания в процессе линейного синтеза и лактамизации; следует отметить, что такие смолы позволяют также осуществлять реакцию при более низких температурах, составляющих 15-25 С, по сравнению со стандартной температурой 40 С, которая требуется для эффективного сочетания, например, на тритиловых смолах. Предпочтительно радикал А (как представлено в формуле II или III) содержит хэндл-фрагмент смолы или линкерный фрагмент смолы, за исключением хэндлов смолы, содержащих аллилоксикарбонильный фрагмент. Более предпочтительно такая смола или хэндл смолы имеет формулу IV в которой R'" обозначает смолу и R"1, R"2, R"3 независимо обозначают водород, C1-С 4 алкил или С 1-С 4 алкоксигруппу и могут иметь одинаковые или различные значения, при условии, что только один из R"1,R"2 может обозначать водород, и где L обозначает кислород, серу, азот или группу формулы V Еще более предпочтительно смола или хэндл смолы имеет формулу VI, в которой радикалы R'",R"1 и R2 имеют указанные выше значения, при условии, что L выбирают из группы, включающей кислород и азот, Еще более предпочтительно смола или хэндл смолы имеет формулу VII, в которой радикалы R"',R"1 и R"2 имеют указанные выше значения, при условии, что L выбирают из группы, включающей кислород и азот,Еще в одном более предпочтительном варианте осуществления изобретения, когда L обозначает кислород, предпочтительно R"1, R"2 независимо обозначают водород, метил или метоксигруппу, при условии, что только один из R"1, R"2 может обозначать водород, и, когда L обозначает азот, указанные радикалы независимо обозначают метил или метоксигруппу, предпочтительно метоксигруппу. Кроме того,еще более предпочтительно L обозначает кислород, R"1 обозначает водород и R2 обозначает метил или метоксигруппу и предпочтительно А обозначает смолу или хэндл смолы. Наиболее предпочтительноR"2 обозначает метил. Наиболее предпочтительно пептид, предлагаемый в настоящем изобретении, связан на С-конце со смолой или хэндлом смолы (А обозначает смолу или хэндл смолы в формуле IV). Предпочтительно последовательность пептида, предлагаемого в настоящем изобретении, представляет собой Ac-Nle-цикло(Asp-His-D-Phe-Arg-Trp-Lys) или Nle-цикло(Asp-His-D-Phe-Arg-Trp-Lys), лактамовая связь находится между боковыми цепями Asp и Lys, как указано в модели пептида и реакционной схеме ниже. Указанный пептид представляет собой обладающий фармацевтической активностью специфический в отношении рецептора меланокортина пептид, который можно применять для лечения сексуальной дисфункции, включая мужскую эректильную дисфункцию и женскую сексуальную дисфункцию у человека. В другом предпочтительном варианте осуществления изобретения последовательность пептида,предлагаемого в настоящем изобретении, представляет собой или содержит, по меньшей мере, частичную последовательность цикло(Asp-His-Phe-Arg-Trp-Lys), в которой остаток Phe может быть также заменен D-Phe или соответствующим D- или L-изомером pF-Phe, Phe(4-Br), Phe(4-CF3), Phe(4-Cl), Phe(2,4 диCl), Phe(3,4-диCl), Phe(3,4-диF), Phe(4-I), Рре(3,4-ди-ОМе), Phe(4-Me) или Phe(4-NO2). Эти модификации с помощью не встречающихся в естественных условиях производных Phe модулируют фармацевтическую активность пептида. Аналогично этому в указанной выше последовательности Arg может быть заменен также D-Arg или соответствующим D- или L-изомером Arg(NO2), Arg(Tos), Arg(Pbf), Arg(Mtr),Arg(Me) или Arg(Pmc). Боковую цепь аргинина предпочтительно можно ковалентно защищать в процессе синтеза, например, с помощью тозила, бензилоксикарбонила, пентаметиленхромансульфонила (Pmc), пентаметилдигидробензофурансульфонила (Pbf), 4-метокси-2,3,6-триметилбензолсульфонила (Mtr) и его 4-трет-бутил 2,3,5,6-тетраметилового гомолога (Tart), адамантилоксикарбонила или Вос. Pmc, Pbf, Mtr или Tart являются очень предпочтительными для защиты Arg, наиболее предпочтительным является Pbf.Trp предпочтительно защищают в процессе синтеза с помощью Boc. Необязательно его можно защищать на N-конце формилом, сим-мезитиленсульфонилом.His предпочтительно защищают N-тритильной защитной группой. Необязательно, хотя это менее предпочтительно, его можно также защищать на N-конце с помощью Boc, метилтритила или тозила. Эксперименты При создании настоящего изобретения синтезировали полностью защищенную с помощью FMOC версию пептида Ac-Nle-цикло(Asp-His-D-Phe-Arg-Trp-Lys), свойства которой были описаны в WO 01/000224, в основном, касательно только указанного пептид, на различных смолах, например на 2-хлортритилполистироловой смоле, и обрабатывали согласно способу, предлагаемому в настоящем изобретении, как указано в модели пептида и реакционной схеме ниже. Используемая на первой стадии реакционной схемы, которая представлена ниже, защищенная на N-конце последовательность представляет собой FMOC-Asp(OAll)-His(Trt)- . Lys(Alloc) и Asp(OAll) представляют собой пять пространственно разделенных остатков, между которыми выступают большие боковые цепи, такие, например, какArg(Pbf). Указанный в нижней части схемы Nle обозначает норлейцин.- 10010786 1.1. Удаление защитных аллильных/Alloc-групп на модели пептида, конъюгированного с 2-CTCсмолой. 0,1 экв. Pd(PPh3)4 солюбилизировали в DCM в присутствии PhSiH3 или диаминборана (5 экв.). Этот раствор добавляли к CTC-смоле, несущей 1 экв. (35 г смолы, загруженность 0,44 ммоля/г) аллил/Alloc-защищенного исходного пептида, представленного в реакционной схеме выше. После проведения реакции в течение максимум 15 мин при комнатной температуре при постоянном барботировании азотом смесь фильтровали и выделенную смолу трижды подвергали той же самой обработке, используя каждый раз такое же количество катализатора и 5 экв. фенилсилана в качестве поглотителя на цикл. Стадия промывки: после этого смолу промывали последовательноNMP (5 раз). 1.2. Циклизация: лактамизация боковых цепей. 1 экв. PyBOP и 1,8 экв. HOBT солюбилизировали в NMP и добавляли к FMOC-защищенному пептиду с незащищенными Asp/Lys в присутствии 4 экв. DIEA. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Лактамизированный пептид выделяли фильтрацией и промывали с помощью NMP. Анализ с помощью ЖХВР с обращенной фазой (колонка С-18, загруженная примерно 0,1 г/мл раствора в 0,1% ТФК 70% воды/30% ацетонитрила, элюирование в градиенте 0,1% ТФК/ацетонитриле) промежуточных образцов пептида, выделенных из CTC-смолы с помощью 2% ТФК, показал, что превращение почти закончено и что 90% лактамизированного защищенного пептида сохраняло FMOC-фрагмент. 1.3. Удаление защитных групп FMOC и удлинение пептида. Удаление защитных групп FMOC осуществляли с помощью 20% пиперидина в NMP. Затем осуществляли удлинение цепи в течение 30 мин в ДМФ примерно при комнатной температуре (40 С) в течение 1 ч с помощью FMOC-L-Nle (1 экв.) в присутствии 1 экв. HCTU и 3 экв. HOOBt, 3 экв. DIEA. После отфильтровывания связанного со смолой продукта и промывки с помощью ДМФ фрагмент FMOC на Nle удаляли с помощью 20% пиперидина в NMP и встраивали N-концевую ацетильную группу путем инкубации в пиридине в присутствии примерно 1,5 экв. ацетангидрида в течение 1-2 ч при комнатной температуре. После отфильтровывания смолы пептид отделяли и осуществляли полное удаление защитных групп путем обработки концентрированной ТФК. Путем грубой оценки устанавливали, что общий выход ацетилированного лактамизированного пептида в пересчете на неочищенный продукт составлял 60%. Необходимость последующей конечной дериватизации на смоле с помощью ацилирующего агента служит дальнейшей демонстрацией ценности настоящего изобретения, а именно для осуществления лактамизации в присутствии лабильной в основных условиях N-защитной группы, но не подвергая ее риску удаления. При использовании отщепляемых с помощью кислоты N-защитных групп, таких как Boc,возникали бы дополнительные проблемы, по меньшей мере, при обработке на смоле. 2.1. Удаление защитных аллильных/Alloc-групп на модели пептида, конъюгированного с бром(4 метилфенил)метилполистироловой смолой, в присутствии аминоборана. В основном, повторяли эксперимент, описанный в разделе 1.1, с использованием (СН 3)2NHBH3 в качестве поглотителя, за исключением того, что CTC-смолу заменяли бром(4-метилфенил)метилполистиролом (примерно 200 меш, 1,2-2,2 ммоля/г) (фирма CBL Patras, Греция). Катализатор солюбилизировали в АсОН/ДМФ (примерно 4:1), в то время как реакцию удаления защитных групп проводили в ДМФ(5-кратный объем по отношению к объему смолы) при 40 С в течение 30 мин. Стадии 2.2 и 2.3 осуществляли аналогично описанным выше стадиям 1.2, 1.3, за исключением того, что отщепление проводили в 5% ТФК в дихлорметане в присутствии 2% TIS. Выход ацетилированного зрелого пептида составлял 72,8% (аналитическая степень чистоты). 3.1. Удаление защитных аллильных/Alloc-групп на модели пептида, конъюгированного с бром(4 метилфенил)метилполистироловой смолой, в присутствии обменного катализатора Pd(Oac)2/P(oTol)3. В основном, повторяли реакцию, описанную в разделе 2.1, но со следующими модификациями: вместо 0,1 экв. Pd(PPh3)4, применяли 0,05 экв. Pd(Oac)2 в присутствии 0,05 экв. ортотолилфосфина. Кроме того, добавляли 2,2 экв. паратолилсульфината натрия в качестве поглотителя. Даже через 2 ч превращение происходило лишь в незначительной степени; повышение температуры до 60 С не изменяло ситуацию. 4.1. Удаление защитных аллильных/Alloc-групп на модели пептида, конъюгированного с бром(4 метилфенил)метилполистироловой смолой, в присутствии обменного катализатора Pd(P[oTol]3)2 и сульфината. Катализатор Pd(P[oTol]3)2 получали согласно методу, описанному у Paul и др., Organometallics, 14(6), 1995, cc. 3030-3039; кроме того, у Paul и др. описано получение родственного соединения Pd(P[2,4- 11010786 ксилоила]3)2. Катализатор солюбилизировали в АсОН/ДМФ (2:1). Реакцию осуществляли практически согласно методу, описанному в разделе 2.1, но с использованием 0,05 экв. Pd(P[oTol]3)2. В качестве поглотителя добавляли 2,2 экв. паратолилсульфината натрия при постоянном барботировании азотом. Реакцию проводили в течение 30 мин при 40 С. Реакция протекала спокойно, после стадии 4.2 аналитический выход отщепленного ацетилированного пептида составлял 82%; стадию 4.3 осуществляли согласно методу, описанному выше в разделе 2. Увеличение времени реакции удаления защитных аллильных/Allocгрупп до 150 мин не приводило к существенному повышению выхода (выход: 84,5%). Уменьшение количества катализатора примерно наполовину существенно замедляло кинетику реакции. 5.1. Удаление защитных аллильных/Alloc-групп на модели пептида, конъюгированного с бром(4 метилфенил)метилполистироловой смолой, в присутствии обменного катализатора Pd(PPh3)4 и сульфината. Реакцию, указанную в разделе 4.1, повторяли практически согласно описанному выше методу, за исключением того, что в качестве катализатора применяли 1 экв. Pd(PPh3)4 и время реакции удаления защитных аллильных/Alloc-групп составляло 30-60 мин. Выход отщепленного ацетилированного зрелого пептида составлял 82%. Предыдущие примеры можно повторять с аналогичным успехом, заменяя описанные в целом или конкретно реагенты и/или рабочие условия для осуществления настоящего изобретения на те, которые применялись в предыдущих примерах. Хотя изобретение было подробно описано с конкретной ссылкой на предпочтительные варианты осуществления изобретения, в других вариантах осуществления изобретения можно достигать таких же результатов. Специалисту в данной области должны быть очевидны вариации и модификации настоящего изобретения, и предполагается, что все такие модификации и эквиваленты подпадают под объем настоящего изобретения. Полные описания всех процитированных ссылок, заявок, патентов и публикаций включены в настоящее описание в качестве ссылки. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ циклизации пептида путем лактамизации, заключающийся в том, что: а) удаляют у пептида защитные группы аллильного типа путем катализа в присутствии Pd(0) и акцептора аллила, где пептид защищен лабильной в основных условиях защитной группой на его N и содержит, кроме того, по меньшей мере одну защищенную аллилоксикарбонилом аминофункцию боковой цепи лизина или аналога такой боковой цепи лизина и, кроме того, содержит по меньшей мере одну защищенную сложным аллиловым эфиром -карбоксильную группу боковой цепи глутамила или аспартила или аналогов таких боковых цепей, при сохранении лабильной в основных условиях защитной группы на N , где аллильные защитные группы являются замещенными или не имеют заместителей,б) осуществляют циклизацию пептида путем лактамизации боковых цепей с удаленными защитными группами в присутствии реагента, представляющего собой слабое основание, и в) удаляют у пептида лабильную в основных условиях защитную группу на его N. 2. Способ по п.1, отличающийся тем, что N, защищенный лабильной в основных условиях защитной группой, представляет собой N аспартильного остатка, имеющего дополнительную защищенную аллиловым сложным эфиром карбоксильную группу, и где предпочтительно аспартильный остаток представляет собой фрагмент дипептидной последовательности Asp-His. 3. Способ по п.1 или 2, отличающийся тем, что на дополнительной конечной стадии г) осуществляют дальнейшее удлинение пептида путем продолжения синтеза пептида или сочетания пептидного фрагмента с N с удаленной защитной группой. 4. Способ по п.3, отличающийся тем, что пептид связывают с твердой фазой в процессе циклизации при условии, что аллильные защитные группы не служат в качестве хэндла смолы и что удлинение пептида на стадии продолжают путем твердофазного синтеза пептида. 5. Способ по п.1 или 2, отличающийся тем, что стадию циклизации б) осуществляют в присутствии реагента, представляющего собой слабое основание, в апротонном полярном растворителе и, кроме того,в присутствии связывающего агента. 6. Способ по п.5, отличающийся тем, что реакцию циклизации осуществляют в присутствии ГХТУ или ТХТУ или фосфониевой соли бензотриазола в качестве связывающего агента. 7. Способ по п.6, отличающийся тем, что пептид связывают со смолой, представляющей собой подложку, с помощью лабильной в кислотных условиях связи. 8. Способ по одному из пп.1-5, отличающийся тем, что пептид ковалентно связывают на С-конце с помощью лабильной в кислотных условиях связи. 9. Способ по п.1, отличающийся тем, что лабильная в основных условиях защитная группа представляет собой группу FMOC. 10. Способ по п.1 или 7, отличающийся тем, что защитные аллильную и Alloc-группы удаляют с помощью катализа с использованием Pd(0) в присутствии акцептора аллила, предпочтительно с использованием избытка R1-PhnSiHm в качестве акцептора аллила, где R1 является заместителем в ароматическом ядре и представляет собой арил, алкил или аралкил, n обозначает 1 или 2 и m обозначает 2 или 3,- 12010786 более предпочтительно где акцептор аллила представляет собой PhSiH3. 11. Циклический пептид формулы II или III, имеющий N, который защищен лабильной в основных условиях защитной группой в которой Y обозначает лабильную в основных условиях защитную группу, n равно 1-10, предпочтительно n равно 1 или 2, наиболее предпочтительно n равно 1, кроме того, в которой m равно 1-15, предпочтительно m равно 3-6, наиболее предпочтительно m равно 3, кроме того, в которой х равно 1-200 и q равно 0-200, предпочтительно х равно 3-50 и q равно 0-50, где R1 и R2, каждый, обозначают встречающуюся в естественных условиях аминокислотную боковую цепь или ее не встречающееся в естественных условиях производное, где боковая цепь может содержать, кроме того, защитную группу, за исключением защитных групп, представляющих собой простой аллиловый эфир и аллилоксикарбонил, и в которой А обозначает смолу или хэндл смолы или в которой R2 необязательно может представлять собой встречающуюся в естественных условиях аминокислотную боковую цепь или ее не встречающееся в естественных условиях производное, где боковая цепь связана со смолой или хэндлом смолы через простой эфир, тиоэфир, сложный эфир, сложный тиоэфир, амидогруппу или фрагмент вторичной или третичной аминогруппы, при условии, что тогда А выбирают из ряда, включающего ОН, NH2, NR'1H илиNR'1R'2, OR'3, где R'1 и R'2 независимо друг от друга обозначают С 1-С 4 алкил. 12. Пептид по п.11, отличающийся тем, что лабильная в основных условиях группа представляет собой FMOC. 13. Пептид по п.11 или 12, отличающийся тем, что пептид имеет формулу II, в которой n обозначает 1 или 2, более предпочтительно n обозначает 1. 14. Пептид по одному из пп.11-13, отличающийся тем, что А представляет собой хэндл смолы или связывающий фрагмент смолы за исключением хэндла смолы, представляющего собой аллилоксикарбонильный фрагмент. 15. Пептид по п.14, отличающийся тем, что смола или хэндл смолы имеет формулу IV в которой R'" обозначает смолу и R"1, R"2, R3 независимо обозначают водород, С 1-С 4 алкил или С 1-С 4 алкоксигруппу и могут иметь одинаковые или различные значения, при условии, что только один из R"1,R"2 может обозначать водород, и где L обозначает кислород, серу, азот или имеет формулу V 16. Пептид по п.15, отличающийся тем, что смола или хэндл смолы имеет формулу VI, в которой- 13010786 радикалы R'", R"1, R2 имеют указанные выше значения, при условии, что L выбирают из группы, включающей кислород и азот, 17. Пептид по п.16, отличающийся тем, что смола или хэндл смолы имеет формулу VII, в которой радикалы R'", R"1, R2 имеют указанные выше значения, при условии, что L выбирают из группы, включающей кислород и азот, 18. Пептид по п.17, отличающийся тем, что когда L обозначает кислород, R"1, R"2 независимо обозначают водород, метил или метоксигруппу, при условии, что только один из R"1, R"2 может обозначать водород и, когда L обозначает азот, независимо обозначают метил или метоксигруппу, предпочтительно метоксигруппу. 19. Пептид по п.18, отличающийся тем, что L обозначает кислород, R"1 обозначает водород, R"2 обозначает метил или метоксигруппу и А предпочтительно обозначает смолу или хэндл смолы. 20. Пептид по п.19, отличающийся тем, что R"2 обозначает метил. 21. Циклический пептид формулы II или III, имеющий N, который защищен лабильной в основных условиях защитной группой в которой Y обозначает лабильную в основных условиях защитную группу, n равно 1-10, предпочтительно n равно 1 или 2, наиболее предпочтительно n равно 1, кроме того, в которой m равно 1-15, предпочтительно m равно 3-6, наиболее предпочтительно m равно 3, кроме того, в которой х равно 1-200 и q равно 0-200, где R1 и R2, каждый независимо, обозначают встречающуюся в естественных условиях аминокислотную боковую цепь или ее не встречающееся в естественных условиях производное, где боковая цепь может содержать, кроме того, защитную группу, за исключением защитных групп, представляющих собой простой аллиловый эфир и аллилоксикарбонил, и в которой А обозначает смолу или хэндл смолы или в которой R2 необязательно представляет собой встречающуюся в естественных ус- 14010786 ловиях аминокислотную боковую цепь или ее не встречающееся в естественных условиях производное,где боковая цепь связана со смолой или хэндлом смолы через простой эфир, тиоэфир, сложный эфир,сложный тиоэфир, амидогруппу или фрагмент вторичной или третичной аминогруппы, при условии, что тогда А представляет собой OR'3, где R'3 обозначает защитную группу, за исключением аллильных групп, предпочтительно R'3 обозначает трет-бутил или пентафторфенил. 22. Применение комплексов Pd(P[орто-толил]3)2 или Pd(P[2,4-ксилоил]3)2 для катализа удаления защищенных аллилом карбоксильных групп, или защищенных аллилоксикарбонилом аминогрупп, и/или гидроксигрупп. 23. Применение по п.22, отличающееся тем, что в качестве акцептора или поглощающего агента аллильной группы используют органический сульфинат. 24. Применение по п.21 или 22, отличающееся тем, что защищенные аллилом или аллилоксикарбонилом группы представляют собой фрагмент необязательно имеющего другие защитные группы пептида, предпочтительно FMOC-защищенного пептида, где группа FMOC связана с N пептида. 25. Способ циклизации пептида путем лактамизации, заключающийся в том, что: а) удаляют у пептида защитные группы аллильного типа путем катализа в присутствии Pd(0) и акцептора аллила, в котором пептид защищен лабильной в основных условиях защитной группой на егоN, который представляет собой N аспартильного остатка, имеющего защищенную сложным аллиловым эфиром -карбоксильную группу и содержит, кроме того, по меньшей мере одну защищенную аллилоксикарбонилом аминофункцию боковой цепи лизина или аналога такой боковой цепи лизина при сохранении лабильной в основных условиях защитной группы на N, где аллильные защитные группы являются замещенными или не имеют заместителей,б) осуществляют циклизацию пептида путем лактамизации боковых цепей с удаленными защитными группами в присутствии реагента, представляющего собой слабое основание, и в) удаляют у пептида лабильную в основных условиях защитную группу на его N.
МПК / Метки
МПК: C07C 29/10, C07K 7/54, C07K 1/06, C07C 51/09, C07C 209/62
Метки: циклизация, пептидов
Код ссылки
<a href="https://eas.patents.su/16-10786-ciklizaciya-peptidov.html" rel="bookmark" title="База патентов Евразийского Союза">Циклизация пептидов</a>
Производные пептидов

Номер патента: 1000
Опубликовано: 28.08.2000
Авторы: Такесима Сатоко, Сакурада Синобу, Нукуи Ерико, Огава Тадаси, Накано Масахару, Таке Нобухиро, Хонго Томоко, Хонго Казуя, Окаяма Тору
МПК: A61K 38/06, C07K 5/087, A61P 29/00...
Метки: пептидов, производные
Формула / Реферат:
1. Соединение, имеющее формулу Y-L-Tyr-Q-NR1-CH(R2)-СО-Х где R1 представляет собой атом водорода; R2 представляет собой незамещенную бензильную группу; Q представляет собой D-Arg; Y представляет собой один атом водорода и C1-6-алкильную группу, или же Y представляет один атом водорода и группу формулы HN=C(R3), где R3 обозначает C1-6-алкильную группу; и Х представляет собой NR7-C(R8)(R9)(R10), где R7 обозначает C1-6-алкильную группу, R8...
Производные пептидов

Номер патента: 1001
Опубликовано: 28.08.2000
Авторы: Огава Тадаси, Сакурада Синобу, Такесима Сатоко, Хонго Казуя, Окаяма Тору, Нукуи Ерико, Накано Масахару, Таке Нобухиро, Хонго Томоко
МПК: A61K 38/06, C07K 5/087, A61P 29/00...
Метки: производные, пептидов
Формула / Реферат:
1. Соединение, представляемое следующей формулой: HN=C (R1)-X-Y-NH-CH(R2)-CO-Q, где R1 представляет собой C1-6-алкильную группу; R2 представляет арилзамещенную C1-6-алкильную группу (предпочтительно фенилзамещенный метил); Х представляет L-тирозин (L-Tyr); Y представляет, по меньшей мере, одну из следующих аминокислот: D-лизин, 2-амино-4-метилсульфинилмасляная кислота (=AMSB или MetO), 2-амино-3-гуанидинпропионовая кислота (=AGPR),...
Способ лечения или профилактики респираторной коронавирусной инфекции с использованием пептидов альфа-тимозина

Номер патента: 9945
Опубликовано: 28.04.2008
Авторы: Рудолф Алфред Р., Татхилл Синтия У.
МПК: A61K 38/00, A61P 31/12
Метки: способ, респираторной, коронавирусной, пептидов, инфекции, профилактики, альфа-тимозина, лечения, использованием
Формула / Реферат:
1. Способ лечения или профилактики респираторной коронавирусной инфекции у больного, включающий введение указанному больному эффективного количества пептида альфа-тимозина. 2. Способ по п.1, в котором указанная респираторная вирусная инфекция представляет собой тяжелый острый респираторный синдром (ТОРС). 3. Способ по п.1, в котором указанное количество пептида альфа-тимозина находится в диапазоне примерно 0,1-20 мг при введении более одного...
Синтез пептидов

Номер патента: 8305
Опубликовано: 27.04.2007
Авторы: Франк Ханс-Георг, Хаберль Удо, Брахт Франц-Петер, Рибка Андреас
МПК: C07K 1/04, C07K 14/55, C07K 1/22...
Формула / Реферат:
1. Применение активированной твердой фазы, содержащей твердый носитель, металл-хелатирующие лиганды, ковалентно связанные с твердым носителем, ионы металла Мn+ с n=1-3, координационно связанные с упомянутыми металл-хелатирующими лигандами, причем указанная активированная твердая фаза обеспечивает координационные участки для координационного и обратимого присоединения якорной части пептида, для твердофазного синтеза пептидов на указанной...
Применение пептидов бактерий streptococcus группы в, кодирующего их полинуклеотида, вакцина и антитело

Номер патента: 9885
Опубликовано: 28.04.2008
Авторы: Эверест Пол, Хенвуд Кэролайн Джоанн, Мур Джоанн Кристин, Добсон Ричард Джеймс, Сантанджело Джозеф Дэвид, Уилсон Ребекка Керри, Лэйн Джонатан Дуглас, Хьюз Мартин Джон Глентон, Дауган Гордон, Фелдман Роберт
МПК: C12N 15/54, C07K 14/315, C12N 15/53...
Метки: пептидов, вакцина, бактерий, группы, антитело, кодирующего, применение, полинуклеотида, streptococcus
Формула / Реферат:
1. Применение пептида орнитинкарбамоилтрансферазы бактерий Streptococcus группы В, кодируемого полинуклеотидом MS4 с последовательностью SEQ ID NO:1, или аналога указанного пептида, обладающего с ним по меньшей мере 70%-ной структурной гомологией и сохраняющего ту же функцию, или их фрагмента для лечения или профилактики инфекции, вызываемой бактериями Streptococcus группы В. 2. Применение пептида орнитинкарбамоилтрансферазы бактерий...
Предыдущий патент: Связывающие молекулы, способные нейтрализовать вирус бешенства, и их применение
Следующий патент: Водный инъекционный раствор антагониста lhrh и способ его получения
Случайный патент: Фунгицидные смеси