Каталитический комплекс и способ получения полиолефинов с мультимодальным молекулярно-массовым распределением
Номер патента: 18934
Опубликовано: 29.11.2013
Авторы: Альт Хельмут, Гёрл Христиан, Палакал Сириак, Абуракабах Атих














Формула / Реферат
1. Трехъядерный каталитический комплекс, содержащий три активных металлических центра, два феноксииминных соединения и два замещенных циклопентадиенильных, инденильных или флуоренильных производных, причем каждое феноксииминное соединение связано с циклопентадиенильным, инденильным или флуоренильным производным, образуя лигандную структуру, циклопентадиенильное, инденильное или флуоренильное производное координировано с одним из металлических центров, а феноксииминное соединение координировано с другим металлическим центром, отличным от металлического центра, с которым координировано циклопентадиенильное, инденильное или флуоренильное производное, где исходное феноксииминное соединение имеет формулу

где R1 представляет собой алкил, C3-C6-циклоалкил или фенил;
R2 представляет собой водород или галоген и
R3 представляет собой C3-C6-циклоалкил или фенил,
где металлический центр выбран из группы, состоящей из солей Ti, Hf и Zr, и
феноксииминное соединение связано с циклопентадиенильным, инденильным или флуоренильным соединением по реакции Соногаширы.
2. Способ получения трехъядерного каталитического комплекса по п.1, включающий стадии связывания замещенного феноксииминного соединения с замещенным циклопентадиенильным, инденильным или флуоренильным производным для получения лигандной структуры и взаимодействия лигандной структуры с активным металлическим компонентом, выбранным из группы, состоящей из солей Ti, Hf и Zr, где феноксииминное соединение связывают с циклопентадиенильным, инденильным или флуоренильным соединением по реакции Соногаширы.
3. Способ полимеризации олефинов, в частности этилена и необязательно одного или более других α-олефинов, отличающийся тем, что в способе применяют каталитический комплекс по п.1 или каталитический комплекс, полученный способом по п.2.
Текст
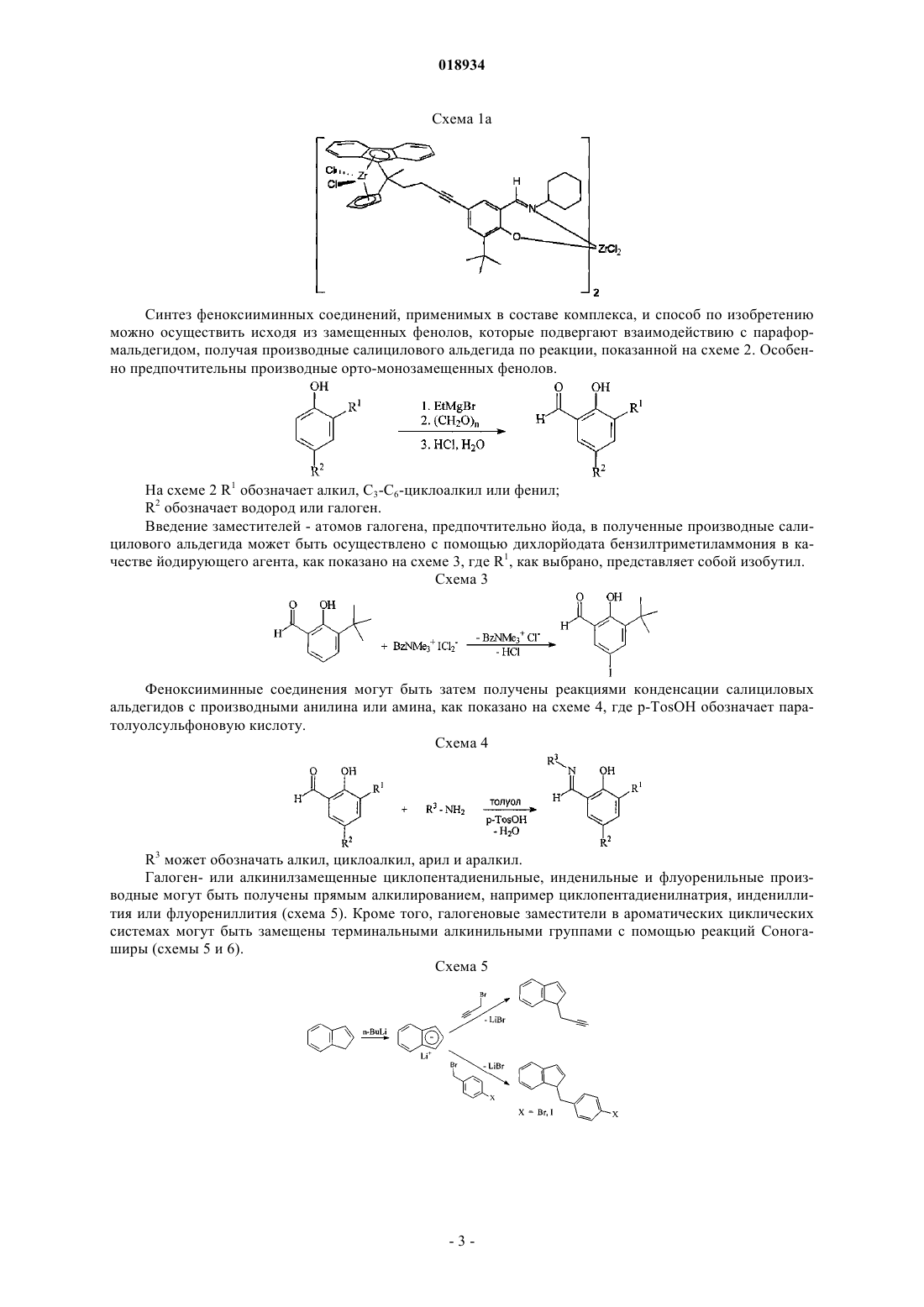
КАТАЛИТИЧЕСКИЙ КОМПЛЕКС И СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВ С МУЛЬТИМОДАЛЬНЫМ МОЛЕКУЛЯРНО-МАССОВЫМ РАСПРЕДЕЛЕНИЕМ В изобретении предложен трехъядерный каталитический комплекс, содержащий три активных металлических центра, два феноксииминных соединения и два замещенных циклопентадиенильных, инденильных или флуоренильных производных, причм каждое феноксииминное соединение связано с циклопентадиенильным, инденильным или флуоренильным производным, образуя лигандную структуру, циклопентадиенильное, инденильное или флуоренильное производное координировано с одним из металлических центров, а феноксииминное соединение координировано с активным металлическим центром, отличным от металлического центра, с которым координировано циклопентадиенильное, инденильное или флуоренильное производное; где феноксииминное соединение имеет формулу(I), где R1 представляет собой алкил, C3-C6-циклоалкил или фенил; R2 представляет собой водород или галоген и R3 представляет собой C3-C6-циклоалкил или фенил; способ получения трехъядерной каталитической композиции,включающий стадии связывания замещенного феноксиимина с замещенным циклопентадиенильным, инденильным или флуоренильным производным для получения лигандной структуры и взаимодействия лигандной структуры с активным металлическим компонентом; и способ полимеризации олефинов, конкретно этилена и одного или нескольких других олефинов, в котором используется указанный трехъядерный комплекс.(71)(73) Заявитель и патентовладелец: САУДИ БЕЙСИК ИНДАСТРИЗ КОРПОРЕЙШН (SA) Настоящее изобретение относится к новой каталитической композиции, которая подходит для получения полиолефинов (ПО) с широким молекулярно-массовым распределением (ММР). Полиолефины с широким молекулярно-массовым распределением (ММР) вызывают интерес из-за своих хороших технологических характеристик. Для получения таких полиолефинов предлагалось множество способов. При первой попытке были получены смеси двух или нескольких ПО, каждый из которых имел конкретную молекулярную массу. Если не проводить очень тщательное смешивание, которое удорожает способ, легко происходит разделение компонентов смеси, что обусловлено различиями в их свойствах, и это затрудняет сохранение постоянного качества полученных продуктов. В еще одном методе было предложено применять последовательно два или несколько реакторов, в каждом из них используются свои особые условия и катализаторы и в каждом получаются ПО, имеющие конкретную молекулярную массу. Смеси продуктов в реакторах также проявляют тенденцию к разделению. Описаны также способы, где в одном реакторе используют два или несколько катализаторов для получения в одну стадию ПО с разными молекулярными массами. Разделение двух полиолефинов с различными молекулярными массами, происходящее в реакторе и после него, является серьезной проблемой. При таком подходе возникает еще одна проблема, которая состоит в том, чтобы найти общие условия полимеризации, в которых все катализаторы проявляют высокую активность. Далее была предложена комбинация катализатора Циглера-Натта и катализатора с единым центром полимеризации, например металлоценового катализатора. Для обоих типов катализаторов обычно используют водород в качестве регулятора цепи для получения ПО с требуемой молекулярной массой. Однако обычно оба типа катализатора требуют различных количеств водорода, что затрудняет получение комбинации с нужной молекулярной массой с приемлемой производительностью. В следующем методе катализатор Циглера-Натта и катализатор с единым центром полимеризации устанавливают на общем носителе, чтобы предотвратить разделение компонентов с различной молекулярной массой мульмодальных ПО. Однако такой подход не решает проблему различных уровней водорода, которые требуются. Кроме того, оказалось, что любой сомономер формирует полимер предпочтительно под действием металлоценового катализатора, что обычно приводит к полимеризации компонентов с более низкой молекулярной массой. Желательно, однако, иметь сомономер, формирующий фракции сополимера с более высокой молекулярной массой, чтобы не допустить появления запаха и дыма во время экструзии полиолефинов. Цель изобретения состоит в создании катализатора, с помощью которого можно получать композиции ПО с широким ММР, которые не разделяются при транспортировке и при других видах обработки. Указанная цель согласно изобретению достигается трехъядерным каталитическим комплексом, содержащим три активных металлических центра, два феноксииминных соединения и два замещенных циклопентадиенильных, инденильных или флуоренильных производных, причем каждое феноксииминное соединение связано с циклопентадиенильным, инденильным или флуоренильным производным, образуя лигандную структуру, причем циклопентадиенильное, инденильное или флуоренильное производное координировано с одним из металлических центров, а феноксииминное соединение координировано с другим активным металлическим центром, отличным от металлического центра, с которым координировано циклопентадиенильное, инденильное или флуоренильное производное, где феноксииминное соединение имеет формулуR2 представляет собой водород или галоген иR3 представляет собой, C3-C6-циклоалкил или фенил,при этом металлический центр выбран из группы, состоящей из солей Ti, Hf, и Zr,и феноксииминное соединение связано с циклопентадиенильным, инденильным или флуоренильным соединением по реакции Соногаширы. Было обнаружено, что каталитический комплекс по изобретению применим для получения полиолефинов с широким молекулярно-массовым распределением (ММР), которые не проявляют тенденции к разделению компонентов с различной молекулярной массой. Значения ММР, составляющие по меньшей мере 15, как было найдено, получаются легко, значения по меньшей мере 20, 30 или даже больше также можно получать. В сополимерах олефинов, полученных с помощью каталитических комплексов по изобретению, сомономеры, как было показано, концентрировались почти полностью в части сополимера с более высокой молекулярной массой. Преимуществом является то, что сополимеры не давали запаха и дыма во время обработки, в частности во время экструзии расплава. Дополнительное преимущество каталитического комплекса по изобретению состоит в том, что не требуется использовать водород во время полимеризации для получения полиолефинов с широким ММР. Если требуется, водород можно добавлять для дальнейшего увеличения ММР полиолефинов, получаемых с помощью катализатора по изобретению. Каталитический комплекс по изобретению содержит два феноксииминных соединения. Феноксиимины координируются с активными металлическими центрами с образованием компонентов катализатора для полимеризации олефинов, в частности этилена. Каталитический комплекс по изобретению содержит два замещенных циклопентадиенильных, инденильных или флуоренильных производных. В дальнейшем подразумевается, что выражение "циклопентадиенильное производное" включает также и инденильное или флуоренильное производное. Циклопентадиенильные производные координируются с активными металлическими центрами с образованием компонентов катализатора для полимеризации олефинов. Металлами, подходящими для образования указанных компонентов катализатора, являются все металлы, про которые известно, что они проявляют каталитическую активность при полимеризации олефинов, когда координированы с феноксииминами и циклопентадиенильными производными. Примерами таких металлов являются Ti, Zr, Hf. Наиболее предпочтительным из них является Zr. Каталитический комплекс по изобретению предпочтительно содержит двухлигандную структуру,где феноксииминные соединения двухлигандной структуры координированы с одним и тем же активным металлическим центром, а циклопентадиенильные производные указанной структуры координированы с разными активными металлическими центрами. Упомянутый комплекс содержит три активных места,одно координировано с феноксииминовой частью двухлигандной структуры, а два координированы с циклопентадиенильными производными, образующими два металлоценовых каталитических центра. Описанный комплекс, как было показано, является очень активным и дает широкое ММР, например по меньшей мере 30. Показано, что предпочтительно поддерживать активные центры, координированные по-разному, в изолированном состоянии, чтобы избежать их взаимного отрицательного влияния, например дать возможность объемным полимерным частицам расти, не препятствуя друг другу. Замещение, по меньшей мере, на феноксииминное или Ср лигандное соединение с -алкинильной группой, по-видимому, представляет собой очень эффективный путь разделения металлических центров. Замещение другого лиганда соответствующим заместителем, например галогеном, предпочтительно бромом или более предпочтительно йодом, делает возможным связывание двух частей лигандов применительно к реакциям сочетания Соногаширы, катализируемым палладием. Кроме того, изобретение относится к способу синтеза многоядерных комплексов переходных металлов, применимых для полимеризации олефинов, в частности этилена, после активации соответствующими сокатализаторами. Конкретно указанный способ представляет собой способ получения многоядерного каталитического комплекса, включающий стадии связывания по меньшей мере одного замещенного феноксииминного соединения по меньшей мере с одним замещенным циклопентадиенильным производным для получения по меньшей мере одной лигандной структуры, взаимодействия по меньшей мере одной лигандной структуры с активным металлическим компонентом путем депротонирования производных феноксиимина и Ср и стадию последующей реакции с солью металла. Полученные комплексы содержат металлические центры с различным окружением, образовавшиеся в результате комбинирования различных предшественников лигандов. Для обеспечения связывания феноксииминного и Ср компонентов они должны содержать заместители, способные реагировать друг с другом. Было показано, что выгодно замещать по меньшей мере один из двух компонентов -алкинильной группой, а другой - галогеном, предпочтительно бромом или йодом, который способен реагировать с алкинильной группой. Взаимодействию с активными металлическими компонентами подвергают предпочтительно структуры с двумя лигандами. Полученный таким образом комплекс, как было найдено, представляет собой такой, как показано на схемах 1 и 1 а. Схема 1 Синтез феноксииминных соединений, применимых в составе комплекса, и способ по изобретению можно осуществить исходя из замещенных фенолов, которые подвергают взаимодействию с параформальдегидом, получая производные салицилового альдегида по реакции, показанной на схеме 2. Особенно предпочтительны производные орто-монозамещенных фенолов.R2 обозначает водород или галоген. Введение заместителей - атомов галогена, предпочтительно йода, в полученные производные салицилового альдегида может быть осуществлено с помощью дихлорйодата бензилтриметиламмония в качестве йодирующего агента, как показано на схеме 3, где R1, как выбрано, представляет собой изобутил. Схема 3 Феноксииминные соединения могут быть затем получены реакциями конденсации салициловых альдегидов с производными анилина или амина, как показано на схеме 4, где p-TosOH обозначает паратолуолсульфоновую кислоту. Схема 4R3 может обозначать алкил, циклоалкил, арил и аралкил. Галоген- или алкинилзамещенные циклопентадиенильные, инденильные и флуоренильные производные могут быть получены прямым алкилированием, например циклопентадиенилнатрия, индениллития или флуорениллития (схема 5). Кроме того, галогеновые заместители в ароматических циклических системах могут быть замещены терминальными алкинильными группами с помощью реакций Соногаширы (схемы 5 и 6). Схема 5 Синтез алкинилзамещенных предшественников мостиковых лигандов металлоценового типа может быть осуществлен реакцией замещенных производных фульвена и циклопентадиенилнатрия, индениллития или флуорениллития (схемы 7 и 8). В качестве исходных соединений для синтеза фульвенов могут быть использованы соответствующим образом замещенные кетоны. Схема 7 Затем осуществляют связывание замещенных соединений феноксиимина и замещенных мостиковых или немостиковых циклопентадиенильных, инденильных или флуоренильных призводных по реакциям сочетания Соногаширы, как показано на схеме 9. Схема 9 Получение трехъядерных каталитических комплексов по изобретению из продуктов реакции сочетания Соногаширы по схеме 9 осуществляют депротонированием предшественников лигандов и последующей реакцией с солью металла. Пример такой реакции показан на схеме 10, где Zr используют в качестве металлических центров. Для получения аналогичных комплексов можно использовать и другие металлы. Ряд каталитических комплексов по изобретению получают и используют в качестве компонентов катализаторов для гомогенной полимеризации и олигомеризации этилена. Комплексы активируют метилалюмоксаном (МАО). Другие соединения бора и алюминия также можно использовать в качестве сокатализаторов. Процессы полимеризации обычно проводят при температурах от 0 до 100C, предпочтительно от 35 до 60C. Применяемое давление этилена предпочтительно составляет от 0,1 до 10 бар. В качестве растворителей можно использовать алканы или ароматические соединения, подобные толуолу. Трехъядерные комплексы дают полиэтилены с бимодальным, мультимодальным или очень широким молекулярно-массовым распределением, что является результатом наличия различных активных центров в молекуле катализатора. Центральная феноксииминная часть дает полиэтилены с низкой или высокой молекулярной массой в зависимости от структуры заместителей. При этом было обнаружено,что циклоалкилзамещенные феноксиимины приводят к полимеру с более низкой молекулярной массой,продуцируемому феноксииминной частью каталитического комплекса, чем фенил- и анилинзамещенные феноксиимины. Молекулярная масса, как было показано, увеличивается при изменении заместителей в следующем порядке: циклопропилциклобутилциклопентилциклогексилфениланилин. Так как возможно также вводить заместители в ароматические циклы металлоценовых частей, которые сильно влияют на молекулярную массу получаемых полиэтиленов, с трехъядерными каталитическими комплексами по изобретению можно получать широкий диапазон полиэтиленов с различными свойствами. Кроме того, изобретение относится к способу полимеризации олефинов, в котором используют катализатор по пункту формулы изобретения 1. Неожиданно было обнаружено, что предложенный способ дает полиолефины с широким ММР, составляющим по меньшей мере 15 и даже более 20 или даже 30. Каталитический комплекс по изобретению можно применять без носителя, но его также можно применять на обычных носителях, например силикагеле, оксиде алюминия. Его можно использовать в известных способах полимеризации олефинов в газовой фазе, суспензии и растворе. Способ подходит для получения гомополимеров полиолефинов, например этилена и пропилена, а также для получения сополимеров этилена с одним или несколькими высшими -олефинами, содержащими от 3 до 10 атомов углерода. В качестве сомономера к этилену предпочтительно используют пропилен, бутен или гексен. Количество используемого сомономера может зависеть от способа получения,требуемых свойств продукта, применяемого катализатора и конкретного сомономера. Упомянутые соображения хорошо известны в данной области техники, и специалист может легко выбрать соответствующий сомономер и его количество, необходимое для получения целевого продукта. Вообще количество используемого сомономера может составлять от 5 до 15 мас.% при производстве HDPE (полиэтилена высокой плотности), а при производстве LLDPE (линейного полиэтилена низкой плотности) оно даже может составлять от 25 до 50 мас.%, считая на общее количество олефиновых мономеров. Изобретение будет пояснено следующими ниже примерами, которые никоим образом его не ограничивают. Пример 1. Синтез 3-трет-бутилсалицилового альдегида (1). К 50 ммоль 2-трет-бутилфенола в 40 мл тетрагидрофурана прибавляют по каплям 3-молярный раствор метилмагнийбромида (18,5 мл; 55,5 ммоль) в диэтиловом эфире. После перемешивания в течение 2 ч при комнатной температуре выделение газа прекращается. Удаляют в вакууме около 90% растворителя и прибавляют толуол (100 мл), триэтиламин (10 мл) и параформальдегид (3,75 г; 125 ммоль). Смесь нагревают до 88C и выдерживают при указанной температуре в течение 2 ч. После охлаждения до комнатной температуры желтый раствор вносят в холодную хлористо-водородную кислоту (1 М, 250 мл). Органическую фазу отделяют и сушат над сульфатом натрия. После удаления растворителя в вакууме 3 трет-бутилсалициловый альдегид очищают перегонкой в высоком вакууме. Выход 88%. Пример 2. Синтез 3-фенилсалицилового альдегида (2). К 100,6 ммоль 2-фенилфенола в 40 мл тетрагидрофурана прибавляют по каплям 3-молярный раствор метилмагнийбромида (36,7 мл; 110 ммоль) в диэтиловом эфире. После перемешивания в течение 2 ч при комнатной температуре выделение газа прекращается. Удаляют в вакууме около 90% растворителя и прибавляют толуол (250 мл), триэтиламин (20 мл) и параформальдегид (7,55 г; 251 ммоль). Смесь нагревают до 88C и выдерживают при указанной температуре в течение 2 ч. После охлаждения до комнатной температуры желтый раствор вносят в холодную хлористо-водородную кислоту (1 М, 250 мл). Органическую фазу отделяют и сушат над сульфатом натрия. После удаления растворителя в вакууме 3 фенилсалициловый альдегид перекристаллизовывают из этанола при -20C. Выход 71%. Пример 3. Синтез 5-хлор-3-циклогексилсалицилового альдегида (3). К 100,4 ммоль 4-хлор-2-циклогексилфенола в 40 мл тетрагидрофурана прибавляют по каплям 3 молярный раствор метилмагнийбромида (36,8 мл; 110,4 ммоль) в диэтиловом эфире. После перемешивания в течение 2 ч при комнатной температуре выделение газа прекращается. Удаляют в вакууме около 90% растворителя и прибавляют толуол (250 мл), триэтиламин (20 мл) и параформальдегид (7,53 г; 251 ммоль). Смесь нагревают до 88C и выдерживают при указанной температуре в течение 2 ч. После охлаждения до комнатной температуры желтый раствор вносят в холодную хлористо-водородную кислоту (1 М, 250 мл). Органическую фазу отделяют и сушат над сульфатом натрия. После удаления растворителя в вакууме 5-хлор-3-циклогексилсалициловый альдегид перекристаллизовывают из этанола при -20C. Выход 59%. Пример 4. Синтез 3-трет-бутил-5-йодсалицилового альдегида (4). 3-трет-Бутилсалициловый альдегид (1) (8,4 ммоль) растворяют в 100 мл смеси метанола и метиленхлорида (3:7). Прибавляют дихлорйодат бензилтриметиламмония (3,23 г; 9,2 ммоль) и безводный карбонат кальция (1,1 г; 11 ммоль). Через 2 ч отфильтровывают избыток карбоната кальция. После удаления около 80% растворителя прибавляют 20 мл раствора гидросульфита натрия (5%), обесцвечивающего смесь. Экстракция диэтиловым эфиром, высушивание над сульфатом натрия и удаление растворителя дают альдегид в виде неочищенного продукта. После очистки перекристаллизацией из н-пентана получают 3-трет-бутил-5-йодсалициловый альдегид с выходом 48% в виде желтых кристаллов, которые слабо чувствительны к свету и их следует хранить в темноте. Пример 5. Синтез N-3-трет-бутилсалицилиден-4-йоданилина (5). 5,84 ммоль 3-трет-Бутилсалицилового альдегида (1) (1,04 г/1 мл) растворяют в 150 мл толуола. После прибавления 4-йоданилина (1,53 г/7 ммоль/1,2 экв.) и нескольких кристаллов паратолуолсульфоновой кислоты реакционную смесь перемешивают с обратным холодильником в течение 3 ч, используя ловушку Дина-Старка. После охлаждения до комнатной температуры прибавляют раствор гидрокарбоната натрия (150 мл). Органическую фазу отделяют и фильтруют через сульфат натрия и силикагель. Удаление растворителя и перекристаллизация из этанола при -20C дают указанное в заголовке соединение с выходом 96%. Пример 6. Синтез N-3-трет-бутилсалицилиден-4-этиниланилина (6). 3-трет-Бутилсалициловый альдегид (1) (0,70 г/3,93 ммоль) и 4-этиниланилин (0,49 г/4,2 ммоль) растворяют в 100 мл этанола. Прибавляют молекулярные сита (3, 15 г) и несколько капель ледяной уксусной кислоты и смесь перемешивают при комнатной температуре в течение трех дней. После фильтрования через сульфат натрия и удаления растворителя остаток несколько раз экстрагируют н-пентаном. Раствор опять фильтруют через сульфат натрия и удаляют растворитель. Получают указанное в заголовке соединение в виде желтого порошка. Выход 43%. С помощью той же реакции, используя толуол в качестве альтернативного растворителя, можно синтезировать другие салицилиденовые производные, например N-3-трет-бутилсалицилиденпропиниламин из 3-трет-бутилсалицилового альдегида и пропиниламина;N-3 фенилсалицилиденпропиниламин из 3-фенилсалицилового альдегида и пропиниламина и N-5-хлор-3 циклогексилсалицилиденпропиниламин из 5-хлор-3-циклогексилсалицилового альдегида и пропиниламина. Пример 7. Синтез N-3-трет-бутил-5-йодсалицилиденциклопентиламина (7). 3-трет-Бутил-5-йодсалициловый альдегид (4) (3,71 г, 5,69 ммоль) растворяют в 150 мл толуола. После прибавления циклопентиламина (0,58 г/6,83 ммоль/1,2 экв.) и нескольких кристаллов паратолуолсульфоновой кислоты реакционную смесь перемешивают при кипячении с обратным холодильником в течение 3 ч, используя ловушку Дина-Старка. После охлаждения до комнатной температуры прибавляют раствор гидрокарбоната натрия (150 мл). Органическую фазу отделяют и фильтруют через сульфат натрия и силикагель. Удаление растворителя приводит к образованию вязкого ярко-желтого масла, которое кристаллизуется при комнатной температуре через несколько дней. Выход 2,10 г (99%). Пример 8. Синтез N-3-трет-бутил-5-йодсалицилиденциклогексиламина (8). 3-трет-Бутил-5-йодсалициловый альдегид (4) (7,64 г, 25,14 ммоль) растворяют в 150 мл толуола. После прибавления циклогексиламина (2,99 г/30,17 ммоль/1,2 экв.) и нескольких кристаллов паратолуолсульфоновой кислоты реакционную смесь перемешивают при кипячении с обратным холодильником в течение 3 ч, используя ловушку Дина-Старка. После охлаждения до комнатной температуры прибавляют раствор гидрокарбоната натрия (150 мл). Органическую фазу отделяют и фильтруют через сульфат натрия и силикагель. Удаление растворителя приводит к образованию вязкого ярко-желтого масла, которое кристаллизуется при комнатной температуре через несколько дней. Выход 9,76 г (98%). Пример 9. Синтез N-3-фенилсалицилиден-4-йоданилина (9). 3-Фенилсалициловый альдегид (2) (4,08 г, 20,6 ммоль) растворяют в 150 мл толуола. После прибавления 4-йоданилина (5,42 г/24,8 ммоль) и нескольких кристаллов пара-толуолсульфоновой кислоты реакционную смесь перемешивают при кипячении с обратным холодильником в течение 3 ч, используя ловушку Дина-Старка. После охлаждения до комнатной температуры прибавляют раствор гидрокарбоната натрия (150 мл). Органическую фазу отделяют и фильтруют через сульфат натрия и силикагель. Удаление растворителя и перекристаллизация из этанола при -20C приводят к образованию указанного в заголовке соединения в виде твердого вещества красного цвета. Выход 7,25 г (88%). Пример 10. Синтез N-5-хлор-3-циклогексилсалицилиден-4-йоданилина (10). 5-Хлор-3-циклогексилсалициловый альдегид (3) (1,90 г, 7,98 ммоль) растворяют в 150 мл толуола. После прибавления 4-йоданилина (2,15 г/9,81 ммоль) и нескольких кристаллов пара-толуолсульфоновой кислоты реакционную смесь перемешивают при кипячении с обратным холодильником в течение 3 ч,используя ловушку Дина-Старка. После охлаждения до комнатной температуры прибавляют раствор гидрокарбоната натрия (150 мл). Органическую фазу отделяют и фильтруют через сульфат натрия и силикагель. Удаление растворителя и перекристаллизация из этанола при -20C приводят к образованию указанного в заголовке соединения в виде твердого вещества оранжевого цвета. Выход 2,95 г (84%). Пример 11. Синтез 5-гексин-2-она (11). 2,4-Пентандион (100 г, 103 мл, 1 моль), безводный карбонат калия (152 г, 1,1 моль) и пропаргилхлорид (71 г, 69 мл, 0,95 моль) растворяют в 500 мл этанола. Реакционную смесь перемешивают при кипячении с обратным холодильником в течение 24 ч. После охлаждения до комнатной температуры прибавляют 300 мл воды. Затем смесь экстрагируют диэтиловым эфиром, органическую фазу промывают насыщенным раствором хлорида натрия и сушат над сульфатом натрия. Удаление растворителя и последующая перегонка в вакууме дают 5-гексин-2-он в виде бесцветной жидкости с выходом 48%. Пример 12. Синтез фульвенового производного 12. Циклопентадиен (125 ммоль) растворяют в 50 мл метанола. Прибавляют 5-гексин-2-он (50 ммоль) и пирролидин (75 ммоль). Смесь перемешивают в течение 24 ч при 40C. В реакционную смесь вводят ледяную уксусную кислоту (100 ммоль), воду (150 мл) и н-пентан (150 мл). Органическую фазу отделяют и сушат над сульфатом натрия. Перегонка в вакууме дает целевое соединение в виде желтого масла (47%). Пример 13. Синтез фульвенового производного 13. Инден (125 ммоль) растворяют в 50 мл метанола. Прибавляют 5-гексин-2-он (50 ммоль) и пирролидин (75 ммоль). Смесь перемешивают в течение четырех дней при 40C. В реакционную смесь вводят ледяную уксусную кислоту (100 ммоль), воду (150 мл) и н-пентан (150 мл). Органическую фазу отделяют и сушат над сульфатом натрия. Перегонка в вакууме дает целевое соединение в виде желтого масла Флуорен (30 ммоль) растворяют в 100 мл диэтилового эфира и подвергают взаимодействию с нбутиллитием (1,6 М, 18,75 мл, 30 ммоль). Через 8 ч прибавляют производное фульвена (12) (30 ммоль) и смесь перемешивают в течение 2 ч при комнатной температуре. Проводят гидролиз, прибавляя 50 мл воды. Отделение органической фазы и удаление растворителя приводят к образованию неочищенного продукта, который перекристаллизовывают из н-пентана при -20C. Выход 73%. Пример 15. Синтез предшественника С 1-мостикового лиганда 15. Инден (12,4 ммоль) растворяют в 100 мл диэтилового эфира и подвергают взаимодействию с нбутиллитием (1,6 М, 7,75 мл, 12,4 ммоль). Через 8 ч прибавляют производное фульвена 13 (12,4 ммоль) и смесь перемешивают в течение 2 ч при комнатной температуре. Проводят гидролиз, прибавляя 50 мл воды. Отделение органической фазы и удаление растворителя приводят к образованию неочищенного продукта, который перекристаллизовывают из н-пентана при -20C. Выход 34%. Пример 16. Синтез предшественника С 1-мостикового лиганда 16. Флуорен (35,1 ммоль) растворяют в 100 мл диэтилового эфира и подвергают взаимодействию с нбутиллитием (1,6 М, 21,95 мл, 35,1 ммоль). Через 8 ч прибавляют производное фульвена 13 (35,1 ммоль) и смесь перемешивают в течение 2 ч при комнатной температуре. Проводят гидролиз, прибавляя 50 мл воды. Отделение органической фазы и удаление растворителя приводят к образованию неочищенного продукта, который перекристаллизовывают из н-пентана при -20C. Выход 47%. Пример 17. Реакция сочетания Соногаширы феноксиимина 8 и предшественника С 1-мостикового лиганда 14. В 15 мл триэтиламина растворяют феноксиимин 8 (1,34 ммоль), предшественник лиганда 14 (1,35 ммоль), дихлорид бис-(трифенилфосфино)палладия (1,3510-5 моль/1 мол.%) и йодид меди(I) (2,710-5 моль/2 мол.%). Смесь перемешивают в течение 20 ч при комнатной температуре. После удаления растворителя прибавляют воду (50 мл) и н-пентан (50 мл). Органическую фазу отделяют и водную фазу экстрагируют несколько раз н-пентаном. Объединенные органические фазы сушат над сульфатом натрия. Удаление растворителя в вакууме, очистка колоночной хроматографией и перекристаллизация из нпентана дают продукт сочетания 17 в виде желтого твердого вещества (выход 94%). Пример 18. Реакция сочетания Соногаширы феноксиимина 8 и предшественника С 1-мостикового лиганда 16. Феноксиимин 8 (1,35 ммоль), предшественник лиганда 16 (1,40 ммоль), дихлорид бис(трифенилфосфино)палладия (1,410-5 моль/1 мол.%) и йодид меди(I) (2,810-5 моль/2 мол.%) растворяют в 15 мл триэтиламина. Смесь перемешивают в течение 20 ч при комнатной температуре. После удаления растворителя прибавляют воду (50 мл) и н-пентан (50 мл). Органическую фазу отделяют и водную фазу экстрагируют несколько раз н-пентаном. Объединенные органические фазы сушат над сульфатом натрия. После удаления растворителя в вакууме, очистки колоночной хроматографией и перекристаллизации из н-пентана получают продукт сочетания 18 в виде оранжевого твердого вещества (выход 81%). Пример 19. Реакция сочетания Соногаширы феноксиимина 7 и предшественника С 1-мостикового лиганда 14. Феноксиимин 7 (1,43 ммоль), предшественник лиганда 14 (1,43 ммоль), дихлорид бис(трифенилфосфино)палладия (1,4310-5 моль/1 мол.%) и йодид меди(I) (2,8610-5 моль/2 мол.%) растворяют в 15 мл триэтиламина. Смесь перемешивают в течение 20 ч при комнатной температуре. После удаления растворителя прибавляют воду (50 мл) и н-пентан (50 мл). Органическую фазу отделяют и водную фазу экстрагируют несколько раз н-пентаном. Объединенные органические фазы сушат над сульфатом натрия. После удаления растворителя в вакууме, очистки колоночной хроматографией и перекристаллизации из н-пентана получают продукт сочетания 19 в виде желтого твердого вещества (выход 86%). Пример 20. Реакция сочетания Соногаширы феноксиимина 9 и предшественника С 1-мостикового лиганда 14. Феноксиимин 9 (1,38 ммоль), предшественник лиганда 14 (1,38 ммоль), дихлорид бис(трифенилфосфино)палладия (1,3810-5 моль/1 мол.%) и йодид меди(I) (2,7610-5 моль/2 мол.%) растворяют в 15 мл триэтиламина. Смесь перемешивают в течение 20 ч при комнатной температуре. После удаления растворителя прибавляют воду (50 мл) и н-пентан (50 мл). Органическую фазу отделяют и водную фазу экстрагируют несколько раз н-пентаном. Объединенные органические фазы сушат над сульфатом натрия. После удаления растворителя в вакууме, очистки колоночной хроматографией и перекристаллизации из н-пентана получают продукт сочетания 20 в виде желтого твердого вещества (выход 68%). Пример 21. Реакция сочетания Соногаширы феноксиимина 10 и предшественника С 1-мостикового лиганда 14. Феноксиимин 10 (1,32 ммоль), предшественник лиганда 14 (1,35 ммоль), дихлорид бис(трифенилфосфино)палладия (1,3510-5 моль/1 мол.%) и йодид меди(I) (2,710-5 моль/2 мол.%) растворяют в 15 мл триэтиламина. Смесь перемешивают в течение 20 ч при комнатной температуре. После удаления растворителя прибавляют воду (50 мл) и н-пентан (50 мл). Органическую фазу отделяют и водную фазу экстрагируют несколько раз н-пентаном. Объединенные органические фазы сушат над сульфатом натрия. После удаления растворителя в вакууме, очистки колоночной хроматографией и перекристаллизации из н-пентана получают продукт сочетания 21 в виде оранжевого твердого вещества (выход 95%). Пример 22. Реакция сочетания Соногаширы феноксиимина 5 и предшественника С 1-мостикового лиганда 14. Феноксиимин 5 (1,50 ммоль), предшественник лиганда 14 (1,51 ммоль), дихлорид бис(трифенилфосфино)палладия (1,5110-5 моль/1 мол.%) и йодид меди(I) (3,0210-5 моль/2 мол.%) растворяют в 15 мл триэтиламина. Смесь перемешивают в течение 20 ч при комнатной температуре. После удаления растворителя прибавляют воду (50 мл) и н-пентан (50 мл). Органическую фазу отделяют и водную фазу экстрагируют несколько раз н-пентаном. Объединенные органические фазы сушат над сульфатом натрия. После удаления растворителя в вакууме, очистки колоночной хроматографией и перекристаллизации из н-пентана получают продукт сочетания 22 в виде желтого твердого вещества (выход 86%). Пример 23. Реакция сочетания Соногаширы феноксиимина 5 и предшественника С 1-мостикового лиганда 15. Феноксиимин 5 (2,51 ммоль), предшественник лиганда 15 (3,0 ммоль), дихлорид бис(трифенилфосфино)палладия (2,5110-5 моль/1 мол.%) и йодид меди(I) (5,0210-5 моль/2 мол.%) растворяют в 15 мл триэтиламина. Смесь перемешивают в течение 20 ч при комнатной температуре. После удаления растворителя прибавляют воду (50 мл) и н-пентан (50 мл). Органическую фазу отделяют и водную фазу экстрагируют несколько раз н-пентаном. Объединенные органические фазы сушат над сульфатом натрия. После удаления растворителя в вакууме, очистки колоночной хроматографией и перекристаллизации из н-пентана получают продукт сочетания 23 в виде светло-коричневого твердого вещества(выход 30%). Пример 24. Реакция сочетания Соногаширы феноксиимина 5 и предшественника С 1-мостикового лиганда 16. Феноксиимин 5 (3,56 ммоль), предшественник лиганда 16 (3,80 ммоль), дихлорид бис(трифенилфосфино)палладия (3,5610-5 моль/1 мол.%) и йодид меди(I) (7,1210-5 моль/2 мол.%) растворяют в 15 мл триэтиламина. Смесь перемешивают в течение 20 ч при комнатной температуре. После удаления растворителя прибавляют воду (50 мл) и н-пентан (50 мл). Органическую фазу отделяют и водную фазу экстрагируют несколько раз н-пентаном. Объединенные органические фазы сушат над сульфатом натрия. После удаления растворителя в вакууме, очистки колоночной хроматографией и перекристаллизации из н-пентана получают продукт сочетания 24 в виде коричневатого твердого вещества (выход 38%). Пример 25. Синтез трехъядерного комплекса из соединения 17. Феноксиимин 17 (0,39 ммоль) растворяют в 30 мл тетрагидрофурана. Прибавляют гидрид калия (47 мг/1,18 ммоль/3 экв.), суспендированный в 10 мл тетрагидрофурана, и смесь перемешивают при комнатной температуре в течение 2 ч до прекращения выделения водорода. Прибавляют тетрахлорид циркония(138 мг/0,59 ммоль/1,5 экв.) и продолжают перемешивание в течение 20 ч. После удаления растворителя в вакууме прибавляют метиленхлорид (30 мл) и раствор фильтруют через сульфат натрия. Удаление примерно 25 мл растворителя в вакууме и последующее прибавление 50 мл н-пентана приводят к осаждению твердого вещества. После декантирования отстоявшегося раствора, промывания н-пентаном (315 мл) и сушки в вакууме получают комплекс 25 в виде оранжевого порошка. Выход 0,18 г (57%). Пример 26. Синтез трехъядерного комплекса из соединения 18. Феноксиимин 18 (0,45 ммоль) растворяют в 30 мл тетрагидрофурана, затем прибавляют нбутиллитий (0,85 мл/1,36 ммоль/3 экв.) в виде 1,6-молярного раствора в смеси гексанов и смесь перемешивают при комнатной температуре в течение 2 ч до прекращения выделения водорода. Прибавляют тетрахлорид циркония (160 мг/0,69 ммоль/1,5 экв.) и продолжают перемешивание в течение еще 20 ч. После удаления растворителя в вакууме прибавляют метиленхлорид (30 мл) и раствор фильтруют через сульфат натрия. Удаление примерно 25 мл растворителя в вакууме и последующее прибавление 50 мл нпентана приводят к осаждению твердого вещества. После декантирования отстоявшегося раствора, промывания н-пентаном (315 мл) и сушки в вакууме получают комплекс 26 в виде коричневатого порошка. Выход 0,21 г (54%). Пример 27. Синтез трехъядерного комплекса из соединения 19. Феноксиимин 19 (0,54 ммоль) растворяют в 30 мл тетрагидрофурана. Прибавляют гидрид калия (66 мг/1,63 ммоль/3 экв.), суспендированный в 10 мл тетрагидрофурана, и смесь перемешивают при комнатной температуре в течение 2 ч до прекращения выделения водорода. Прибавляют тетрахлорид циркония(190 мг/0,82 ммоль/1,5 экв.) и продолжают перемешивание в течение 20 ч. После удаления растворителя в вакууме прибавляют метиленхлорид (30 мл) и раствор фильтруют через сульфат натрия. Удаление примерно 25 мл растворителя в вакууме и последующее прибавление 50 мл н-пентана приводят к осаждению твердого вещества. После декантирования отстоявшегося раствора, промывания н-пентаном (315 мл) и сушки в вакууме получают комплекс 27 в виде зеленовато-желтого порошка. Выход 0,41 г (95%). Пример 28. Синтез трехъядерного комплекса из соединения 20. Феноксиимин 20 (0,43 ммоль) растворяют в 30 мл тетрагидрофурана. Прибавляют гидрид калия (52 мг/1,29 ммоль/3 экв.), суспендированный в 10 мл тетрагидрофурана, и смесь перемешивают при комнатной температуре в течение 2 ч до прекращения выделения водорода. Прибавляют тетрахлорид циркония(150 мг/0,65 ммоль/1,5 экв.) и продолжают перемешивание в течение 20 ч. После удаления растворителя в вакууме прибавляют метиленхлорид (30 мл) и раствор фильтруют через сульфат натрия. Удаление примерно 25 мл растворителя в вакууме и последующее прибавление 50 мл н-пентана приводят к осаждению твердого вещества. После декантирования отстоявшегося раствора, промывания н-пентаном (315 мл) и сушки в вакууме получают комплекс 28 в виде коричневатого порошка. Выход 0,31 г (88%). Пример 29. Синтез трехъядерного комплекса из соединения 21. Феноксиимин 21 (0,36 ммоль) растворяют в 30 мл тетрагидрофурана. Прибавляют гидрид калия (43 мг/1,07 ммоль/3 экв.), суспендированный в 10 мл тетрагидрофурана, и смесь перемешивают при комнатной температуре в течение 2 ч до прекращения выделения водорода. Прибавляют тетрахлорид циркония(124 мг/0,54 ммоль/1,5 экв.) и продолжают перемешивание в течение 20 ч. После удаления растворителя в вакууме прибавляют метиленхлорид (30 мл) и раствор фильтруют через сульфат натрия. Удаление примерно 25 мл растворителя в вакууме и последующее прибавление 50 мл н-пентана приводят к осаждению твердого вещества. После декантирования отстоявшегося раствора, промывания н-пентаном (315 мл) и сушки в вакууме получают комплекс 29 в виде коричневатого порошка. Выход 0,30 г (96%). Пример 30. Синтез трехъядерного комплекса из соединения 22. Феноксииминное соединение 22 (0,44 ммоль) растворяют в 30 мл тетрагидрофурана. Прибавляют гидрид калия (53 мг/1,32 ммоль/3 экв.), суспендированный в 10 мл тетрагидрофурана, и смесь перемешивают при комнатной температуре в течение 2 ч до прекращения выделения водорода. Прибавляют тетрахлорид циркония (154 мг/0,66 ммоль/1,5 экв.) и продолжают перемешивание в течение 20 ч. После удаления растворителя в вакууме прибавляют метиленхлорид (30 мл) и раствор фильтруют через сульфат натрия. Удаление примерно 25 мл растворителя в вакууме и последующее прибавление 50 мл н-пентана приводят к осаждению твердого вещества. После декантирования отстоявшегося раствора, промывания н-пентаном (315 мл) и сушки в вакууме получают комплекс 30 в виде желтого порошка. Выход 0,37 г(95%). Пример 31. Синтез трехъядерного комплекса из соединения 23. Феноксиимин 23 (0,45 ммоль) растворяют в 30 мл тетрагидрофурана. Прибавляют гидрид калия (54 мг/1,34 ммоль/3 экв.), суспендированный в 10 мл тетрагидрофурана, и смесь перемешивают при комнатной температуре в течение 2 ч до прекращения выделения водорода. Прибавляют тетрахлорид циркония(157 мг/0,68 ммоль/1,5 экв.) и продолжают перемешивание в течение 20 ч. После удаления растворителя в вакууме прибавляют метиленхлорид (30 мл) и раствор фильтруют через сульфат натрия. Удаление примерно 25 мл растворителя в вакууме и последующее прибавление 50 мл н-пентана приводят к осаж- 12018934 дению твердого вещества. После декантирования отстоявшегося раствора, промывания н-пентаном (315 мл) и сушки в вакууме получают комплекс 31 в виде коричневатого порошка. Выход 0,14 г (39%). Пример 32. Синтез трехъядерного комплекса из соединения 24. Феноксиимин 24 (0,38 ммоль) растворяют в 30 мл тетрагидрофурана. Прибавляют гидрид калия (48 мг/1,20 ммоль/3 экв.), суспендированный в 10 мл тетрагидрофурана, и смесь перемешивают при комнатной температуре в течение 2 ч до прекращения выделения водорода. Прибавляют тетрахлорид циркония(133 мг/0,57 ммоль/1,5 экв.) и продолжают перемешивание в течение 20 ч. После удаления растворителя в вакууме прибавляют метиленхлорид (30 мл) и раствор фильтруют через сульфат натрия. Удаление примерно 25 мл растворителя в вакууме и последующее прибавление 50 мл н-пентана приводят к осаждению твердого вещества. После декантирования отстоявшегося раствора, промывания н-пентаном (315 мл) и сушки в вакууме получают комплекс 32 в виде коричневатого порошка. Выход 0,11 г (34%). Пример 33. Общая методика полимеризации этилена в автоклаве Bchi объемом 1 л. Целевой комплекс (1-10 мг) суспендируют в 5 мл толуола. Смесь переносят в колбу Шленка объемом 1 л, заполненную 250 мл н-пентана. Полученную смесь переносят в лабораторный автоклав от Bchi объемом 1 л в инертной атмосфере. Этилен под давлением 1 бар подают в течение 5 мин. Метилалюмоксан (30% в толуоле, Zr:Al = 1:500) прибавляют с помощью канюли, автоклав термостатируют (35C), подавая этилен под давлением 10 бар в течение более 1 ч. Полученный полимер отфильтровывают на стеклянном фильтре, промывают разбавленной соляной кислотой, водой, ацетоном и в заключение сушат в вакууме. Свойства полиэтилена, полученного с различными каталитическими комплексами, показаны в таблице. Результаты реакций полимеризации этилена ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Трехъядерный каталитический комплекс, содержащий три активных металлических центра, два феноксииминных соединения и два замещенных циклопентадиенильных, инденильных или флуоренильных производных, причем каждое феноксииминное соединение связано с циклопентадиенильным, инденильным или флуоренильным производным, образуя лигандную структуру, циклопентадиенильное, инденильное или флуоренильное производное координировано с одним из металлических центров, а феноксииминное соединение координировано с другим металлическим центром, отличным от металлического центра, с которым координировано циклопентадиенильное, инденильное или флуоренильное производное, где исходное феноксииминное соединение имеет формулуR2 представляет собой водород или галоген иR3 представляет собой C3-C6-циклоалкил или фенил,где металлический центр выбран из группы, состоящей из солей Ti, Hf и Zr, и феноксииминное соединение связано с циклопентадиенильным, инденильным или флуоренильным соединением по реакции Соногаширы. 2. Способ получения трехъядерного каталитического комплекса по п.1, включающий стадии связы- 13018934 вания замещенного феноксииминного соединения с замещенным циклопентадиенильным, инденильным или флуоренильным производным для получения лигандной структуры и взаимодействия лигандной структуры с активным металлическим компонентом, выбранным из группы, состоящей из солей Ti, Hf иZr, где феноксииминное соединение связывают с циклопентадиенильным, инденильным или флуоренильным соединением по реакции Соногаширы. 3. Способ полимеризации олефинов, в частности этилена и необязательно одного или более других-олефинов, отличающийся тем, что в способе применяют каталитический комплекс по п.1 или каталитический комплекс, полученный способом по п.2.
МПК / Метки
МПК: C07F 17/00, C08F 10/00, B01J 31/22, C08F 4/659, B01J 31/18
Метки: получения, комплекс, молекулярно-массовым, каталитический, полиолефинов, распределением, способ, мультимодальным
Код ссылки
<a href="https://eas.patents.su/15-18934-kataliticheskijj-kompleks-i-sposob-polucheniya-poliolefinov-s-multimodalnym-molekulyarno-massovym-raspredeleniem.html" rel="bookmark" title="База патентов Евразийского Союза">Каталитический комплекс и способ получения полиолефинов с мультимодальным молекулярно-массовым распределением</a>
Способ получения полимера, обладающего бимодальным молекулярно-массовым распределением, способ модификации молекулярно-массового распределения статистического сополимера изобутилена, полимерный продукт и полимерная композиция на его основе

Номер патента: 1222
Опубликовано: 25.12.2000
Автор: Уайт Дональд Эндрю
МПК: C08F 8/00
Метки: основе, молекулярно-массовым, молекулярно-массового, модификации, сополимера, композиция, бимодальным, продукт, полимера, распределением, обладающего, получения, полимерный, распределения, изобутилена, способ, полимерная, статистического
Формула / Реферат:
1. Способ получения полимера, обладающего бимодальным молекулярно-массовым распределением, из полимера, обладающего мономодальным молекулярно-массовым распределением, молекулярная масса которого уменьшается при перемешивании с высокой сдвиговой деформацией необязательно в присутствии инициатора свободнорадикальной полимеризации, характеризующийся тем, что исходный полимер выбирают из группы, включающей полипропилен, сополимеры пропилена и...
Каталитическая композиция для получения полиолефинов, имеющих би- или мультимодальное молекулярно-массовое распределение, активная каталитическая система и способ ее получения, способ полимеризации олефинов и применение активной каталитической системы для получения линейного полиэтилена

Номер патента: 17068
Опубликовано: 28.09.2012
Автор: Разави Аббас
МПК: C08F 4/646, C08F 10/00, C08F 4/70...
Метки: композиция, активная, полимеризации, линейного, способ, распределение, применение, би, олефинов, молекулярно-массовое, мультимодальное, полиолефинов, система, полиэтилена, получения, каталитической, системы, активной, имеющих, каталитическая
Формула / Реферат:
1. Каталитическая композиция для получения полиолефинов, имеющих би- или мультимодальное молекулярно-массовое распределение, которая включает:а) по меньшей мере один металлоценовый каталитический компонент формулы (I)где Cp является замещенным или незамещенным циклопентадиенильным кольцом;C'p является замещенным или незамещенным флуоренильным кольцом;R" является структурным мостиком между Cp и С'р, придающим компоненту пространственную...
Способ получения полиолефинов

Номер патента: 15021
Опубликовано: 29.04.2011
Авторы: Корхонен Эса, Карбаси Амир Киумарс, Бергстра Михил
МПК: C08F 2/14, C08F 10/00
Метки: способ, полиолефинов, получения
Формула / Реферат:
1. Способ получения полиолефинов, который включает следующие стадии:полимеризацию по меньшей мере одного олефинового мономера;отделение по крайней мере части реакционной смеси;разделение выделенной реакционной смеси на обедненную полимером фракцию и обогащенную полимером фракцию;при этом по меньшей мере часть обедненной полимером фракции подвергают очистке перед тем, как вновь возвратить ее в реакцию полимеризации по меньшей мере одного...
Каталитический способ получения гидропероксидов алкилбензолов посредством аэробного окисления в мягких условиях

Номер патента: 18658
Опубликовано: 30.09.2013
Авторы: Рекуперо Франческо, Порта Омбретта, Гамбаротти Кристиан, Миниши Франческо, Пунта Карло, Спаччини Раффаэле
МПК: C07C 407/00, C07C 409/08
Метки: гидропероксидов, алкилбензолов, посредством, условиях, получения, мягких, аэробного, каталитический, окисления, способ
Формула / Реферат:
1. Способ получения гидропероксидов алкилбензолов, выбранных из группы, состоящей из этилбензола, втор-бутилбензола и фенилциклогексана, отличающийся тем, что алкилбензол реагирует с кислородом в присутствии каталитической системы, состоящей из N-гидроксиимида в сочетании с пероксидным активатором, имеющим структуру перкислоты, где количество указанного активатора составляет от 0,1 до 5 мол.% в пересчете на количество алкилбензола.2. Способ по...
Твердая или полутвердая композиция, содержащая молекулярно диспергированный дроспиренон, и способ ее получения

Номер патента: 12570
Опубликовано: 30.10.2009
Авторы: Вагнер Торстен, Функе Адриан
МПК: A61K 31/565, A61K 9/00, A61K 9/107...
Метки: полутвердая, молекулярно, диспергированный, содержащая, твердая, способ, композиция, дроспиренон, получения
Формула / Реферат:
1. Твердая или полутвердая композиция для перорального введения, содержащая дроспиренон, молекулярно диспергированный по крайней мере в одном фармацевтически приемлемом носителе. 2. Композиция по п.1 в твердой форме, в которой по крайней мере один из фармацевтически приемлемых носителей является твердым при комнатной температуре и/или точка плавления которого находится в диапазоне от 40 до 80шС и выбран из группы, состоящей из полиэтиленгликоля...
Предыдущий патент: Производное бензодиазепина для лечения гематопоэтических новообразований и лейкоза
Следующий патент: Нуклеозидный ингибитор для вгс (hcv)
Случайный патент: Способ и установка для выделения органических веществ из содержащей их газовой смеси