Пирометаллургический способ обработки металлсодержащих материалов










Формула / Реферат
1. Способ концентрирования вольфрама из вольфрамсодержащего материала, включающий следующие этапы:
(a) нагрев вольфрамсодержащего материала в присутствии неорганического соединения щелочного металла и при практически полном отсутствии галоидной соли при температуре, достаточной для расплавления упомянутого вольфрамсодержащего материала в течение времени, достаточного для образования фазы с большей плотностью и фазы с меньшей плотностью, причем большая часть упомянутого вольфрама концентрируется в фазе с большей плотностью в виде соли вольфрама,
(b) разделение упомянутой фазы с большей плотностью и упомянутой фазы с меньшей плотностью под действием силы тяжести, причем упомянутая фаза с большей плотностью оседает на дно,
(c) концентрирование упомянутого вольфрама путем отделения упомянутой фазы с большей плотностью от фазы с меньшей плотностью,
(d) получение карбида вольфрама путем барботирования упомянутой фазы с большей плотностью углеродсодержащим газом при повышенной температуре с целью получения карбида вольфрама, и
(e) рециклирование части солесодержащего материала из барботированной фазы с большей плотностью этапа (d) в расплав, содержащий вольфрамсодержащий материал, в присутствии соединения щелочного металла упомянутого выше в этапе (а).
2. Способ по п.1, в котором упомянутый вольфрамсодержащий материал выбирают из группы, состоящей из гюбнерита (MnWO4), шеелита (CaWO4), ферберита (FeWO4), вольфрамита ((Fe,Mn)WO4) и их смесей.
3. Способ по п.1, в котором упомянутый вольфрамсодержащий материал выбирают из группы, состоящей из колошниковой пыли, шлаков, лома и их смесей.
4. Способ по п.1, в котором упомянутое соединение щелочного металла выбирают из группы, состоящей из соединений натрия и соединений калия.
5. Способ по п.1, в котором упомянутое соединение щелочного металла выбирают из группы, состоящей из силиката натрия, карбоната натрия, гидроксида натрия и их смесей.
6. Способ по п.1, в котором этап нагревания (а) производят при температуре от приблизительно 900шС до приблизительно 1200шС.
7. Способ по п.1, в котором этап нагревания (а) производят при температуре от приблизительно 1050шС до приблизительно 1150шС.
8. Способ по п.1, в котором этап барботирования (d) производят при температуре от приблизительно 1050шС до приблизительно 1200шС.
9. Способ по п.1, в котором этап барботирования (d) производят при температуре от приблизительно 1050шС до приблизительно 1150шС.
10. Способ по п.1, в котором упомянутым углеродсодержащим газом является газообразный углеводород.
11. Способ по п.1, в котором упомянутый углеродсодержащий газ выбирают из группы, состоящей из метана, этилена, пропана и их смесей.
12. Способ по п.1, в котором упомянутую фазу с высокой плотностью отделяют от упомянутой фазы с низкой плотностью путем сливания упомянутой фазы с высокой плотностью из тигля.
13. Способ по п.1, в котором упомянутую фазу с высокой плотностью удаляют через отверстие в тигле, содержащем упомянутую фазу с высокой плотностью и упомянутую фазу с низкой плотностью.
14. Способ по п.1, в котором упомянутый карбид вольфрама, образованный на этапе (d), отделяют от остального материала и очищают.
15. Способ по п.14, в котором упомянутый карбид вольфрама имеет чистоту не менее 90% после очистки.
16. Способ получения карбида вольфрама, включающий следующие этапы:
(a) нагрев концентрата вольфрамсодержащего минерала в присутствии неорганического соединения натрия или калия при практически полном отсутствии хлорида натрия до температуры от приблизительно 900шС до приблизительно 1200шС для получения первичного расплава,
(b) поддержание упомянутого первичного расплава при температуре от приблизительно 900шС до приблизительно 1200шС до тех пор, пока упомянутый расплав не разделится на вольфрамсодержащую фазу с большей плотностью и шлаковую фазу с меньшей плотностью, причем, по меньшей мере, 80% вольфрама в упомянутом вольфрамовом минеральном концентрате концентрируется в упомянутой вольфрамсодержащей фазе с большей плотностью,
(c) отделение упомянутой вольфрамсодержащей фазы с большей плотностью от шлаковой фазы с меньшей плотностью,
(d) нагрев упомянутой вольфрамсодержащей фазы с большей плотностью до температуры от приблизительно 1050шС до приблизительно 1200шС для получения вторичного расплава,
(e) пропускание газообразного метана через упомянутый вторичный расплав для получения барботированного вторичного расплава, содержащего карбид вольфрама,
(f) отделение части расплава, обогащенной карбидом вольфрама, от упомянутого барботированного вторичного расплава, и
(g) очистка упомянутой части расплава, обогащенной карбидом вольфрама, для получения очищенного карбида вольфрама.
17. Способ по п.16, в котором часть солесодержащего материала берут из упомянутого вторичного расплава и помещают (рециклируют) в первичный расплав типа упомянутого в этапе (а) для участия в отделении упомянутой вольфрамсодержащей фазы с большей плотностью от шлаковой фазы с меньшей плотностью.
18. Способ по п.16, в котором упомянутая вольфрамсодержащая фаза с большей плотностью содержит соль оксида вольфрама.
19. Способ по п.16, в котором упомянутая вольфрамсодержащая фаза с большей плотностью содержит соль вольфрамата натрия.
20. Способ по п.16, в котором упомянутая шлаковая фаза с меньшей плотностью содержит силикат.
21. Способ по п.16, в котором упомянутая шлаковая фаза с меньшей плотностью содержит силикат марганца, силикат железа или силикаты алюминия-кальция.
22. Способ по п.16, в котором упомянутый карбид вольфрама образуется в виде мелких частиц, причем не менее 90% частиц карбида вольфрама имеют средний диаметр менее 10 мкм.
23. Способ по п.16, в котором не менее 95% вольфрама в упомянутом вольфрамсодержащем минерале концентрируется в упомянутой вольфрамсодержащей фазе с большей плотностью.
24. Способ по п.16, в котором не менее 97% вольфрама в упомянутом вольфрамсодержащем минерале концентрируется в упомянутой вольфрамсодержащей фазе с большей плотностью.
25. Способ по п.16, в котором, по крайней мере, часть упомянутого соединения натрия выбирают из группы, состоящей из: (i) силиката натрия (Nа2SiO3) или (ii) карбоната (Nа2СО3) в присутствии кремнезема (SiO2).
26. Способ по п.16, в котором упомянутый этап очистки (g) включает:
(a) сухое дробление, воздушную сепарацию и сухую сепарацию упомянутой части расплава, обогащенной карбидом вольфрама,
(b) водное выщелачивание упомянутого очищенного сухим способом материала,
(c) разделение прошедшего водное выщелачивание материала на твердую и жидкую фазы для получения кристаллов карбида вольфрама,
(d) дробление и кислотное выщелачивание упомянутых кристаллов неочищенного карбида вольфрама, и
(e) разделение прошедшего дробление и кислотное выщелачивание кристаллического неочищенного карбида вольфрама на твердую и жидкую фазы для получения высокочистого карбида вольфрама.
27. Способ по п.16, в котором упомянутый очищенный карбид вольфрама имеет содержание карбида вольфрама не менее 90%.
28. Способ получения карбида вольфрама, включающий следующие этапы:
(а) нагрев соли вольфрамовой кислоты до температуры, превосходящей температуру ее плавления, для получения расплава при практически полном отсутствии каких-либо галоидных солей, разделение расплава на фазу с большей плотностью и фазу с меньшей плотностью, причем большая часть вольфрама концентрируется в упомянутой фазе с большей плотностью,
(b) пропускание газообразного углеводорода через упомянутую фазу с большей плотностью для получения карбида вольфрама, и
(c) отделение упомянутого карбида вольфрама от остальной фазы с большей плотностью.
29. Способ по п.28, в котором упомянутой солью вольфрамовой кислоты является вольфрамат натрия.
30. Способ получения карбида вольфрама, включающий следующие этапы:
(a) нагрев концентрртр вольфрамсодержащего минерала в присутствии неорганического соединения натрия или калия при практически полном отсутствии хлорида натрия до температуры от приблизительно 900шС до приблизительно 1200шС для получения первичного расплава,
(b) поддержание упомянутого первичного расплава при температуре от приблизительно 900шС до приблизительно 1200шС до тех пор, пока упомянутый расплав не разделится на вольфрамсодержащую фазу с большей плотностью и шлаковую фазу с меньшей плотностью,
(c) отделение упомянутой вольфрамсодержащей фазы с большей плотностью от шлаковой фазы с меньшей плотностью,
(d) нагрев упомянутой вольфрамсодержащей фазы с большей плотностью до температуры от приблизительно 1050шС до приблизительно 1200шС для получения вторичного расплава,
(e) пропускание газообразного метана через упомянутый вторичный расплав для получения барботированного вторичного расплава, содержащего карбид вольфрама,
(f) отделение части расплава, обогащенной карбидом вольфрама, от упомянутого барботированного вторичного расплава, и
(g) очистка упомянутой части расплава, обогащенной карбидом вольфрама, для получения очищенного карбида вольфрама.
31. Способ по п.5, в котором упомянутый солесодержащий материал, подвергаемый рециклированию, представляет собой материал, выбранный из группы, состоящей из вольфрамата натрия, Na2O и их смесей.
32. Способ по п.17, в котором упомянутый рециклируемый солесодержащий материал представляет собой материал, выбранный из группы, состоящей из вольфрамата натрия, Na2O и их смесей.
33. Способ по п.29, в котором часть упомянутой оставшейся фазы с большей плотностью этапа (е) рециклируют в расплав типа упомянутого в этапе (а).
34. Способ по п.26, в котором жидкую фазу, отделяемую на этапе (с), подают в кристаллизатор, а кристаллы рециклируют в упомянутый вторичный расплав.
Текст
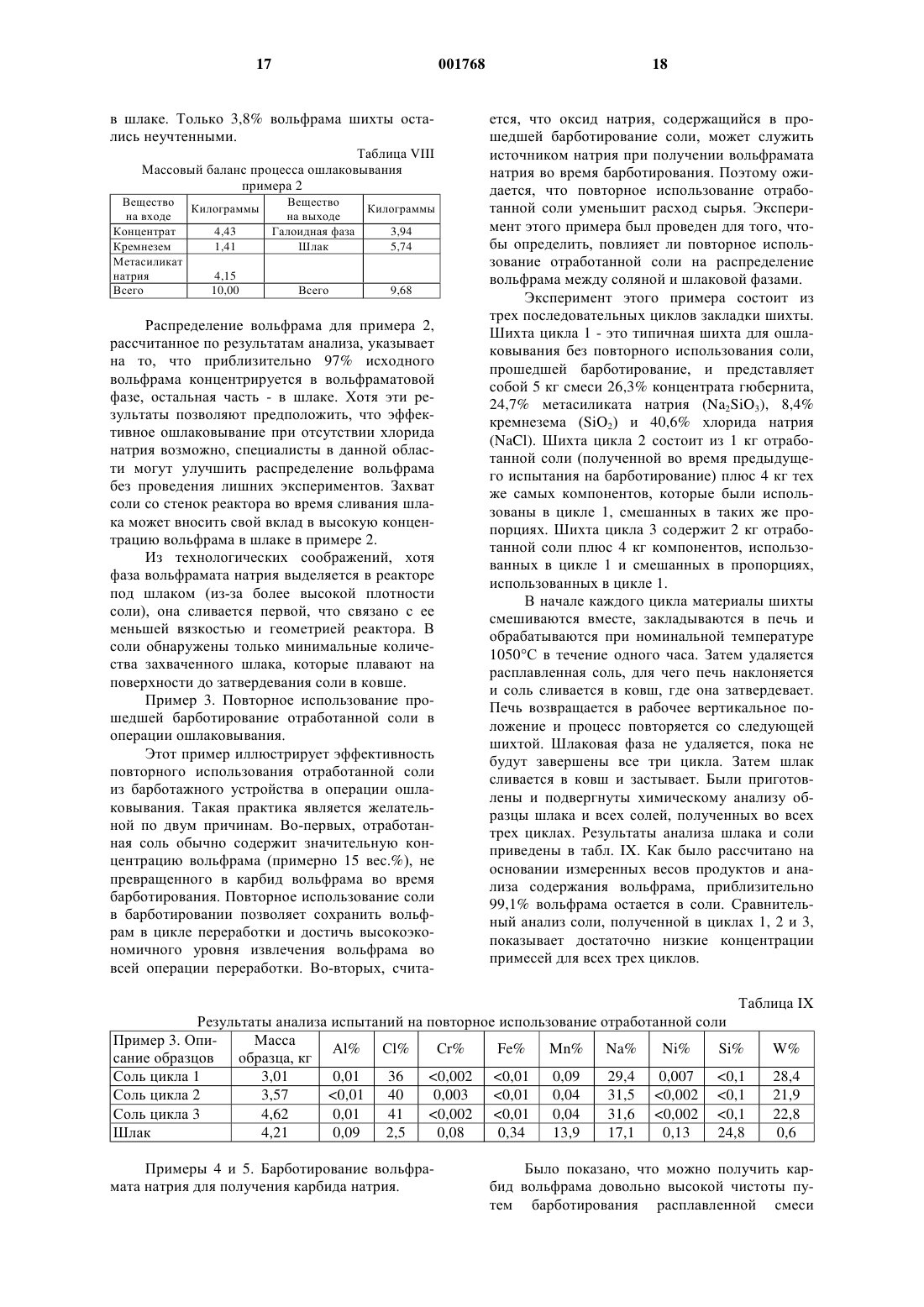
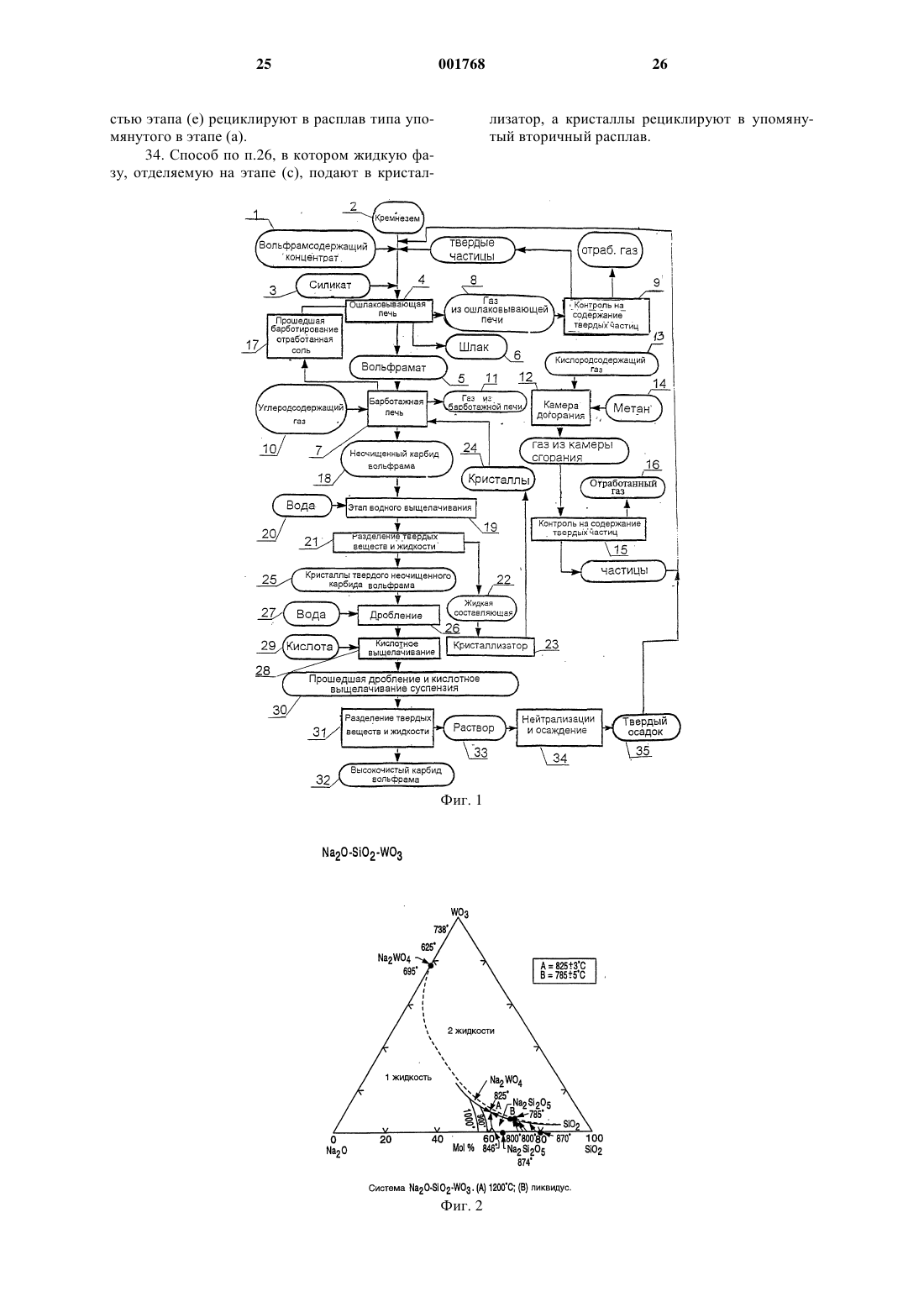
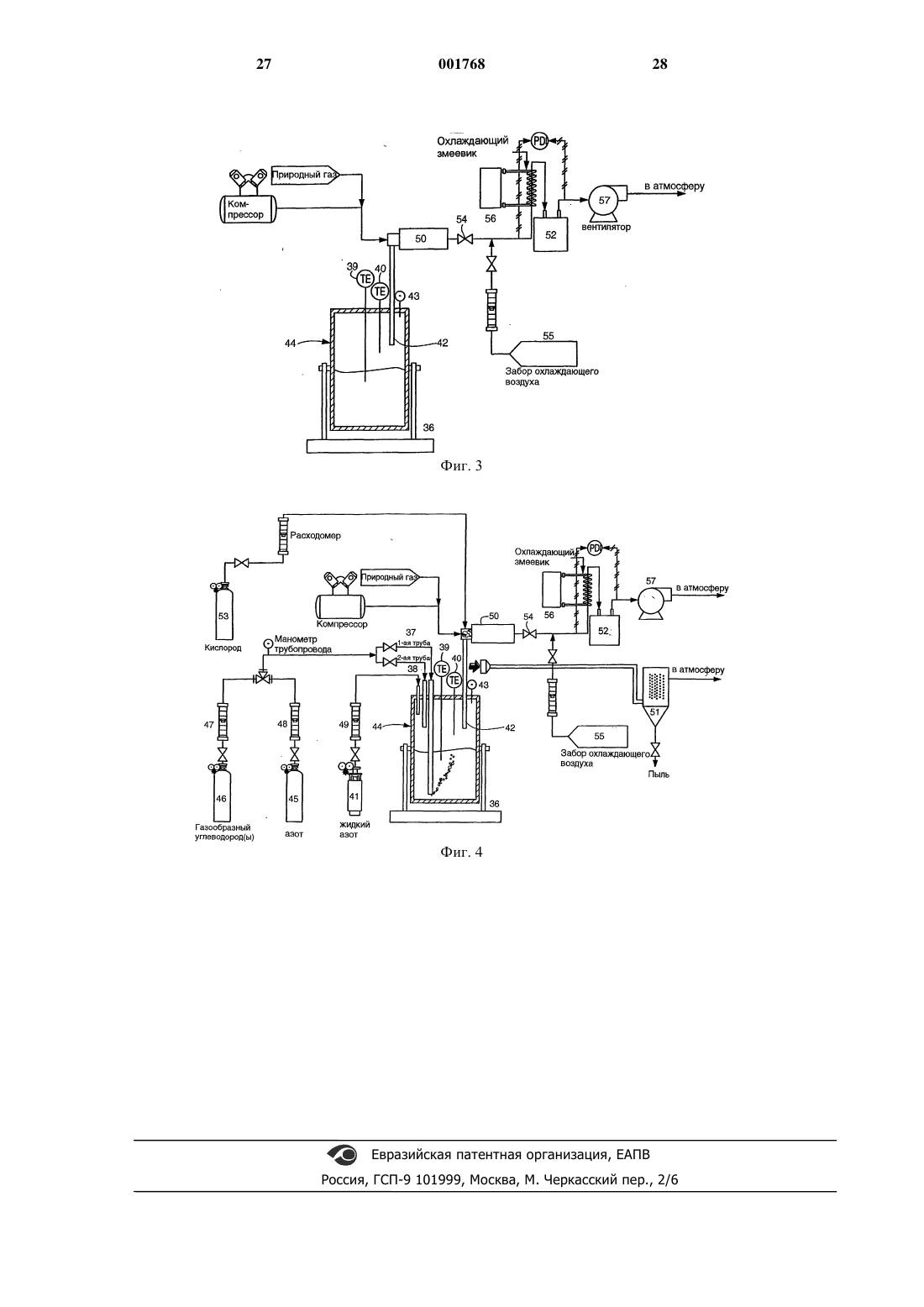
1 Область, к которой относится изобретение Настоящее изобретение относится к пирометаллургической обработке металлсодержащих материалов и в предпочтительном варианте осуществления к получению карбида вольфрама с использованием двухступенчатого пирометаллургического способа. Предпосылки создания изобретения Известны двухступенчатые способы получения карбида вольфрама (WC). Например, в патенте США 3.373.097 под названием Способ отделения металлсодержащей галоидной фазы из силикатной фазы с содержанием породы и электролиз галоидной фазы с целью получения металла Gomes и др., выданном 12 марта 1968 г., описан способ получения карбида вольфрама. Способ включает в себя отделение расплавленной фазы с использованием хлорида натрия (NaCl), при котором вольфрам в основном концентрируется в менее плотной верхней галоидной фазе, тогда как примесные элементы,такие как кальций, марганец и железо собираются в более плотной нижней силикатной фазе. На процесс отделения влияет нагрев смеси галоидных солей, концентратов шеелита (CaWO4) или вольфрамита Fe,Mn)WO4), и вещества,формирующего шлаки, такого как силикат натрия, до температуры от 900 до 1100 С. После выдержки от пятнадцати минут до часа при повышенной температуре процесс разделения фаз завершается, и галоидная фаза сливается для обработки путем электролиза расплавленной соли. Патент США 4.489.044 под названием Получение монокарбида вольфрама из расплавленной вольфрамо-галоидной фазы способом газового барботажа Gomes и др., выданный 18 декабря 1984 г., переизданный как Re 32/612 23 февраля 1988 г., описывает способ получения карбида вольфрама. Способ включает в себя получение фазы хлорида натрия/вольфрамата натрия (Na2WO4) путем разделения расплавленных фаз и подобен описанному выше способу. Монокарбид вольфрама получается путем барботирования расплава хлорида натрия и вольфрамата натрия углеводородным газом, в частности метаном (СН 4) или природным газом. В соответствии с описанием изобретения хлорид натрия может быть замещен другими щелочными галоидами. В мае 1985 г. Gomez, Raddatz и Caranahan сделали доклад на Третьем симпозиуме по вольфраму в Мадриде, Испания (13-17 мая 1985 г.), касающийся двухэтапной технологии получения гранулярного порошка карбида вольфрама непосредственно из концентратов шеелита или вольфрамита. Сначала концентраты вступали в реакцию с расплавом хлорида натрия/метасиликата натрия (Na2SiO3) при температуре 1050 С. В результате реакции получались две несмешивающиеся жидкости: более легкая вольфрамо-галоидная фаза (NaCl 001768Na2WO4), содержащая 99% исходного вольфрама, и более плотная фаза силикатного шлака,содержащая от 90 до 96% железа, марганца и оксидов кальция. После разделения фаз вольфрамо-галоидная фаза на втором этапе подвергалась барботированию газообразным метаном для получения гранулярного карбида вольфрама. Карбид вольфрама извлекается из реактора путем слива излишка соли, охлаждения, водного выщелачивания и соскабливания. См. Получение карбида вольфрама газовым барботированием расплавов вольфрамата" Gomez и др.,Journal of Metals, декабрь 1985, стр. 29-32. Все описанные выше процессы включают в себя этап ошлаковывания, на котором концентрат вольфрама смешивается с кремнеземистым плавнем и хлоридом натрия (могут быть использованы и другие источники галоидов). Соединения вольфрама, содержащиеся в концентрате (например, вольфраматы кальция, железа или марганца) реагируют с хлоридом натрия и силикатом натрия и образуют две несмешивающиеся фазы: расплавленную соль и расплавленный силикатный шлак. Вольфрам в основном концентрируется в фазе расплавленной соли, тогда как большинство примесей попадают в шлаковую фазу. Вязкий шлак имеет большую плотность, чем соль, и осаждается на дно тигля печи. Соляная фаза, которая в основном состоит из хлорида натрия и вольфрамата натрия, используется на втором этапе процесса для получения карбида вольфрама. С описанными выше способами связана проблема, которая заключается в том, что вольфрамсодержащая фаза, имеющая меньшую плотность, также включает в себя галоидную соль (например, хлорид натрия). Во время последующей операции барботирования эта галоидная соль испаряется и осаждается внутри различных деталей системы подачи газа. Такое осаждение соли периодически приводит к простоям, связанным с необходимостью очистки системы. Необходимость удаления хлорида натрия также приводит к повышению эксплуатационных расходов. Кроме того, хлорид натрия чрезвычайно коррозиен, и его присутствие повышает стоимость используемых материалов из-за необходимости использовать коррозионно-устойчивые материалы и повышает эксплуатационные расходы из-за наличия коррозии. Кроме того, хлорид натрия растворяет вольфрамат натрия во время барботирования, что приводит к уменьшению химической активности оксида вольфрама (WO3). Было бы желательно иметь способ получения карбида металла (например, карбида вольфрама) из металлсодержащего минерала с использованием пирометаллургического способа. Кроме того, было бы желательно получать карбид металла (например, вольфрама) без необходимости получения расплавленной металлогалоидной соли. Было бы желательно иметь спо 3 соб, в котором большая часть вольфрама, участвующего в процессе, преобразовывалась бы в карбид вольфрама. Было бы желательно иметь способ, посредством которого можно было бы эффективно и экономично получать карбид вольфрама без длительных простоев системы. Краткое изложение сущности изобретения В соответствии с одним из вариантов осуществления настоящего изобретения предлагается способ концентрирования металла в металлсодержащих веществах, в котором используется пирометаллургический способ. Пирометаллургический способ включает в себя этап нагревания, на котором металлсодержащее вещество нагревается в присутствии, по крайней мере, одного натриевого или калиевого соединения для расплавления металлсодержащего вещества и образования металлсодержащей фазы с высокой плотностью и шлаковой фазы с низкой плотностью. Большая часть металла концентрируется в металлсодержащей фазе с высокой плотностью. Эти две фазы не смешиваются, и металлсодержащая фаза с высокой плотностью отделяется от шлаковой фазы с низкой плотностью за счет силы тяжести. Из-за своей более высокой плотности металлсодержащая фаза с высокой плотностью осаждается на дне тигля печи. После этого эти две фазы можно разделить. Желательно подвергнуть металлсодержащую фазу с высокой плотностью вторичной пирометаллургической обработке,т.е. барботированию с использованием углеродсодержащего газа, для получения карбида металла. Хотя и было обнаружено, что способы настоящего изобретения в основном применимы к вольфрамсодержащим материалам, эти способы также могут быть применены для выделения других металлов из металлсодержащих материалов. Примерами таких других металлов могут служить металлы группы III-B (например,торий), металлы группы IV-B (например, титан,цирконий, гафний), металлы группы V-B (например, ванадий, ниобий, тантал), металлы группы VI-B (например, вольфрам) и металлы группы VII-B (например, марганец и рений). Более предпочтительными являются тугоплавкие металлы, такие как вольфрам, титан и тантал. Наиболее предпочтительными являются вольфрамсодержащие материалы. Примерами вольфрамсодержащих материалов могут служить вольфрамовые руды, такие как гюбнерит(MnWO4), шеелит (CaWO4), ферберит (FeWO4) и вольфрамит Fe,Mn)WO4). Кроме того, способ настоящего изобретения может эффективно применяться и в случае использования других вольфрамсодержащих материалов, таких как колошниковая пыль и различное вторсырье (например, шлак и лом). Хотя способы настоящего изобретения пригодны для целого ряда материалов, для простоты изложения ниже приведено описание предпочтительного варианта осу 001768 4 ществления, в котором используются вольфрамсодержащие материалы. Следует ясно понимать, что можно также использовать и другие материалы, такие как перечисленные выше. В соответствии с другим вариантом осуществления настоящего изобретения карбид вольфрама получается из концентрата вольфрамового минерала. Концентрат вольфрамового минерала нагревается в присутствии натриевого или калиевого соединения до температуры от приблизительно 900 С до приблизительно 1200 С для получения первичного расплава. Первичный расплав поддерживается при этой температуре до тех пор, пока он не разделится на вольфрамсодержащую фазу с высокой плотностью и шлаковую фазу с низкой плотностью. Затем вольфрамсодержащая фаза с высокой плотностью отделяется от шлаковой фазы с низкой плотностью. Вольфрамсодержащая фаза с высокой плотностью нагревается до температуры от приблизительно 1050 С до приблизительно 1200 С для получения вторичного расплава. Затем газообразный метан пропускается через вторичный расплав для получения карбида вольфрама. Обогащенная карбидом вольфрама часть вторичного расплава удаляется и очищается для получения очищенного карбида вольфрама. Желательно, чтобы первый расплав образовывался при практическом отсутствии хлорида натрия. В предпочтительном варианте осуществления изобретения часть прошедшего барботирование отработанного солесодержащего материала поступает из вторичного расплава в первичный расплав, чтобы способствовать отделению вольфрамсодержащей фазы с высокой плотностью от шлаковой фазы с низкой плотностью и для извлечения вольфрама, не превращенного в карбид вольфрама на этапе барботирования. Краткое описание чертежей На фиг. 1 приведена схема последовательности операций одного из вариантов осуществления способа настоящего изобретения; на фиг. 2 - экспериментально полученная фазовая диаграмма системы WO3 - Na2O - SiO2 при 1200 С; на фиг. 3 - схема экспериментальной установки для ошлаковывания в соответствии с одним из вариантов осуществления настоящего изобретения; на фиг. 4 - схема экспериментальной установки для барботирования в соответствии с одним из вариантов осуществления настоящего изобретения. Подробное описание изобретения В соответствии с одним из вариантов осуществления настоящего изобретения предлагается пирометаллургический способ получения вольфраматовой соли, например вольфрамата натрия или вольфрамата калия, из вольфрамсодержащего материала. Желательно, чтобы вольфрамсодержащим материалом была вольф 5 рамовая руда, такая как гюбнерит (MnWO4),шеелит (CaWO4), ферберит (FeWO4) и вольфрамит Fe,Mn)WO4) или вольфрамсодержащие материалы, такие как колошниковая пыль и различное вторсырье (например, шлак и лом). Способ пирометаллургического шлакования включает в себя нагрев вольфрамсодержащего материала в присутствии шлакообразующего силиката (предпочтительно кремнезема и силиката щелочного металла). Расплав разделяется на две несмешивающиеся фазы, более плотную вольфрамсодержащую фазу и менее плотную шлаковую фазу. В соответствии с другим вариантом осуществления настоящего изобретения предлагается способ получения карбида вольфрама из вольфрамсодержащего материала. Желательно,чтобы способ включал в себя два пирометаллургических этапа, первый этап шлакования и второй этап барботирования. На фиг. 1 приведена схема последовательности операций предпочтительного варианта осуществления способа настоящего изобретения. Вольфрамсодержащий концентрат 1 вместе с кремнеземом 2 и силикатом натрия 3 помещаются в ошлаковывающую печь 4. В ошлаковывающей печи 4 поддерживается температура в диапазоне от приблизительно 900 до приблизительно 1200 С, предпочтительно от приблизительно 1050 до приблизительно 1150 С, более предпочтительно приблизительно 1050 С в течение приблизительно 0,5-2,0 ч. Помещенные в печь материалы разделяются на две несмешиваемые фазы. Вольфрамсодержащая фаза с более высокой плотностью (вольфрамат) 5 осаждается на дне тигля печи под действием силы тяжести, и менее плотная шлаковая фаза (силикат) 6 сегрегируется в верхней части тигля печи. Более плотная вольфрамсодержащая фаза 5 помещается в барботажную печь 7. Шлаковая фаза с меньшей плотностью 6 может быть удалена в отходы или подвергнута дальнейшей переработке. Вольфрамсодержащая фаза с большей плотностью 5 может быть отделена от шлаковой фазы с меньшей плотностью 6 с помощью целого ряда соответствующих способов. Например,фаза с большей плотностью 5 и фаза с меньшей плотностью 6 могут быть слиты последовательно через устье наклоняющейся или вращающейся печи в соответствующие емкости, например ковши. С другой стороны, в тигле может быть предусмотрено сливное отверстие для слива вольфрамсодержащей фазы 5 с более высокой плотностью. Газ 8 из ошлаковывающей печи 4 может быть подвергнут контролю на содержание твердых частиц 9. Выделенные твердые вещества могут быть помещены на переработку в ошлаковывающую печь 4, отработанный газ может быть выброшен в атмосферу. Вольфрамсодержащая фаза 5 с более высокой плотностью помещается в барботажную печь 7 и нагревается до температуры в диапазо 001768 6 не от приблизительно 1050 до приблизительно 1200 С, предпочтительно от приблизительно 1050 до приблизительно 1150 С и более предпочтительно до температуры приблизительно 1100 С. Углеродсодержащий газ 10, например метан, вводится в барботажную печь 7. Углеродсодержащий газ 10 разлагается при температуре барботажной печи, и образуется углерод,необходимый для получения карбида вольфрама. Газ 11 из барботажной печи 7 может подаваться в камеру догорания 12 с добавлением кислородсодержащего газа, например, воздуха 13 и углеводорода, например, метана 14. Газ в камере догорания может пройти контроль на содержание твердых частиц 15. Извлеченные твердые вещества могут быть снова помещены в ошлаковывающую печь 4, а отработанный газ 16 может быть выброшен в атмосферу. Прошедшая барботирование отработанная соль 17 может быть снова помещена в ошлаковывающую печь 4. После этапа барботирования получается неочищенный карбид вольфрама 18, который представляет собой серый спеченный материал. Неочищенный карбид вольфрама 18 подвергается водному выщелачиванию 19 после добавления воды 20, после чего следует разделение твердых веществ и жидкости 21. Жидкая составляющая 22 подается в кристаллизатор 23, и кристаллы 24 могут быть снова помещены в барботажную печь 7. Кристаллы твердого неочищенного карбида вольфрама 25 дробятся 26 в воде 27 и подвергаются кислотному выщелачиванию 28 с использованием подходящей кислоты (например, НСl). В соответствии с предпочтительным вариантом осуществления настоящего изобретения дробление 26 и кислотное выщелачивание 28 происходят в одном процессе. Дробление 26 предпочтительно производится в шаровой мельнице с использованием абразивного материала из карбида вольфрама. Сначала приготавливается взвесь неочищенных кристаллов карбида вольфрама 25 в разбавленном водном растворе соляной кислоты 29, и дробление 26 производится в течение времени,достаточного, чтобы высвободить и перевести примеси в твердое состояние. Прошедшая дробление и кислотное выщелачивание суспензия 30 подвергается разделению жидкой и твердой фаз 31. Твердый высокочистый карбид вольфрама 32 предпочтительно имеет степень чистоты не менее 90% карбида вольфрама, более предпочтительно не менее 95% карбида вольфрама и более предпочтительно не менее 99% карбида вольфрама. Раствор 33 подвергается нейтрализации и осаждению 34 твердых веществ 35. Твердый осадок 35 после сушки может быть снова помещен в ошлаковывающую печь 4. Во время первой пирометаллургической операции содержимое печи, представляющее собой смесь концентрата вольфрама и кремнеземистого плавня, обрабатывается при темпера 7 туре приблизительно 1050 С. Вольфрамовые соединения, содержащиеся в концентрате (например, вольфраматы кальция, железа или марганца), реагируют с кремнеземистым плавнем(предпочтительно силикатом натрия и кремнеземом) и образуют две несмешиваемые фазы: расплавленную соль и расплавленный силикатный шлак. Вольфрам в основном концентрируется в фазе расплавленной соли, а большинство примесей удаляются в шлак. Соль имеет большую плотность, чем шлак, и осаждается на дне тигля печи. Соляная фаза, которая в основном состоит из вольфрамата натрия, затем подвергается второму этапу пирометаллургической обработки, т.е. барботированию. Концентрат, используемый в примерах,содержит гюбнерит (MnWO4) в качестве первичного вольфрамсодержащего минерала. Когда смесь претерпевает описанную выше обработку,происходит следующая химическая реакция:MnWO4(c)+Na2SiO3(c)=MnOSiO2(1)+Na2WO4(1) Разделение соляной и шлаковой фаз становится возможным благодаря существованию области несмешиваемости, которой обладает система оксид вольфрама - оксид натрия - кремнезем, как показано на фиг. 2. Когда концентраты вольфрама попадают в расплав при температуре 1050 С, они реагируют с силикатом натрия с образованием вольфрамата натрия и шлака. При этой температуре шлак и вольфрамат не смешиваются и разделяются за счет силы тяжести. Точный химический состав шлака будет зависеть от относительного содержания избыточного кремнезема и оксида натрия в системе. Оксид натрия является желательной составляющей шлака, так как его наличие понижает точку плавления шлака достаточно, чтобы обеспечить образование полностью жидкой фазы. При отсутствии оксида натрия или другого плавня, эффективно понижающего точку плавления шлака, невозможно получить жидкий шлак в системе оксид марганца - кремнезем при температурах ниже приблизительно 1250 С. Пример ошлаковывающей установки приведен на фиг. 3. За исключением системы нагнетания газа, та же самая базовая конфигурация печи, которая используется в операции барботирования (описанной ниже), может быть использована и для ошлаковывания. Так как операция ошлаковывания представляет собой просто расплавление и разделение фаз, никакие газовые трубки или магистрали для продувки азота не нужны. Второй пирометаллургический процесс способствует кристаллизации карбида вольфрама в расплавленной соляной фазе. Эта цель достигается путем нагрева вольфрамсодержащей расплавленной соли, полученной на первом этапе процесса, до температуры, лежащей в диапазоне приблизительно от 1080 до 1100 С с последующим барботированием газообразным 8 углеводородом, таким как метан или пропан,при большом стехиометрическом избытке газа. В таких условиях газообразный углеводород разлагается и обеспечивает восстановитель и углерод, необходимые для получения карбида вольфрама. Фаза карбида вольфрама представляет собой кристаллы микронного размера, которые не растворяются в фазе расплавленной соли. Кристаллы также имеют большую плотность, чем соль, и концентрируются около дна реактора. После завершения барботирования отработанная соль сливается с кристаллов. Когда в качестве источника углеводорода используется метан, то предполагается, что конечной чистой химической реакцией, приводящей к образованию карбида вольфрама (WC),является 4 CH4(g)+Na2WO4(1)=WC(s)+3 CO(g)+8 H2(g)+Na2O(1) Так как необходим значительный стехиометрический избыток газообразного углеводорода, некоторая часть газа также разлагается на углерод и газообразный водород в соответствии со следующей реакцией:CH4(g)=C(s)+2 H2(g) Газообразный водород и большая часть свободного углерода окисляются в камере догорания. Однако некоторая часть углерода остается в соляной фазе в качестве загрязняющей примеси. Таким образом, желательно свести к минимуму образование избыточного углерода в барботажной печи. Вместо метана можно использовать другие газообразные углеводороды, такие как пропан и этилен (С 2 Н 4). Например, использование этилена может повысить эффективность барботирования (т.е. повысить выход карбида вольфрама на единицу углерода, добавляемую в расплав). После отделения свободно текущей фазы отработанной соли, образовавшиеся кристаллы карбида вольфрама остаются в отдельной фазе серого цвета, имеющей вид спеченного вещества. Это серое вещество содержит значительное количество соли. Соль и другие примеси удаляются в процессе сухого или мокрого дробления и последующего выщелачивания в соляной кислоте, каустической соде и воде. После такой обработки получившиеся в результате кристаллы могут содержать от 99,3 до 99,4% карбида вольфрама. Однако карбид вольфрама, полученный в результате предварительных испытаний, имеет существенно более высокую концентрацию примесей. Предполагается, что повышенная концентрация примесей, в основном хрома и никеля, является результатом химического воздействия расплавленной соли в реакционном тигле. Приведенная в качестве примера система,пригодная для ошлаковывания (фиг. 3) и барботирования (фиг. 4), включает в себя наклоняющуюся печь 36 с крышкой, которая вмещает в себя две трубы для барботирования 37, 38, две термопары 39, 40, одну специальную трубу для 9 подачи азота 41, одну линию вывода 42 и один манометр 43. Основным компонентом является печь 36 с резистивным нагревом на 12,9 кВт. Печь 36 имеет зону нагрева длиной 0,914 м и диаметром 12,7 см; она позволяет достигать максимальной рабочей температуры 1200 С. Внутри кожуха печи установлен реактор или тигель 44, изготовленный из трубы Inconel 600 диаметром 10,2 см; максимальная глубина бака составляет 45,7 см. Тигель 44 можно вынимать для очистки или обслуживания после открывания снабженного петлями кожуха печи. Одна и та же печь может быть использована для ошлаковывания (фиг. 3) и барботирования (фиг. 4). Как в операции ошлаковывания,так и в операции барботирования исходная шихта обычно помещается в холодный тигель 44, после чего на печь 36 подается питание и температура бака поднимается до нужной величины. Последующие загрузки могут производиться в горячую печь 36. Чтобы облегчить извлечение расплавленных материалов, печь 36 можно отклонять на угол до 180 от вертикального рабочего положения для слива материалов в ковши. Для работы в режиме газового барботирования (фиг. 4) была сконструирована система,способная продувать тигель 44 азотом 45 и барботировать расплавленный материал метаном,пропаном, этиленом или любой смесью этих газообразных углеводородов 46. Во время барботирования газовые трубы 37, 38 пропускаются через два отверстия. Газообразные углеводороды 46 нагнетаются в тигель 44 через одну из труб 37, а вторая труба 38 является резервной на случай засорения первой трубы 37. Желательно,чтобы трубы 37, 38 имели сравнительно маленький внутренний диаметр (например, 0,14 см) для обеспечения относительно высокой скорости потока для сведения к минимуму разложения газа в трубах 37, 38. Газообразные углеводороды 46 нагнетаются непосредственно в расплав в точке, расположенной примерно на 5 см выше дна тигля 44. Во время всех барботажных испытаний через третье входное отверстие в точку, расположенную приблизительно на 2,5 см ниже крышки реактора, также нагнетается азот по трубе 41. Наличие специальной трубы для подачи азота 41 обеспечивает положительное давление в пространстве над расплавом в печи, что не позволяет воздуху попадать в тигель 44. Все входящие газовые потоки контролируются расходомерами 47, 48, 49. Остальные три отверстия в крышке служат для ввода термопар, а основное (центральное) отверстие служит для отвода отработанного газа через линию вывода 42. Основной задачей при конструировании системы было обеспечить эффективную транспортировку выделяющегося углерода, который образуется при разложении или как продукт барботажной реакции, в камеру догорания 50. В 10 одном варианте осуществления изобретения давление внутри печи 36 сначала устанавливается на уровне от 0,25 до 0,50 миллиметров (мм) водяного столба путем выравнивания потока реагирующего газа с тягой вытяжки. Затем давление повышается до уровня от 2,5 до 5,0 мм водяного столба путем регулировки потока в специальной трубе подачи азота 41. Отметим,что конец этой трубы 41 расположен на расстоянии всего около 5 см от верха тигля 44, так что большая часть тяги расходуется на удаление газов, являющихся продуктами реакции, а не азота. Таким образом, большая часть азота растекается по поверхности верхней крышки, поддерживая инертную атмосферу в верхней части тигля 44 и не давая воздуху контактировать с образованными в результате реакции газами внутри печи 36. Образованные в результате реакции газы вытягиваются в камеру догорания 50 и сжигаются с образованием Н 2 О и СО 2. Захваченная соль может быть собрана в фильтре 51 или газоочистителе 52. Когда используется фильтр 51, газы проходят через мокрый газоочиститель; затем они выбрасываются в атмосферу. В варианте осуществления изобретения,изображенном на фиг. 4, выходящий из камеры догорания 50 газ проходит через газоочиститель 52, а фильтр 51 используется для обработки возможных выбросов загрязнений из печи 36. Технологические газы выходят из тигля 44 через единственную линию вывода 42 и поступают в систему обработки газа. Газы поступают непосредственно в камеру догорания 50, изготовленную из нержавеющей стали, имеющую диаметр 20,3 см и длину 55,9 см, работающую на природном газе, по трубе диаметром 5 см. В камере догорания 50 используется находящаяся под давлением растопочная горелка, которая горит в течение всего времени проведения испытаний. Кислород 53 в контролируемом количестве подается в камеру догорания 50 для сжигания выделенного водорода, остатков газообразных углеводородов и мелких частиц углерода, попавших в поток выходящего газа. После выхода из камеры догорания 50 газы проходят через шаровой клапан 54 (используется для балансировки профиля давления газа в системе) и затем поступают в газоочиститель 52. Газоочиститель 52 состоит из 208 литрового полимерного бака и системы поливинилхлоридных (ПВХ) труб. Газоочиститель наполнен приблизительно 114 л воды; выходящие из камеры догорания газы в виде пузырьков попадают в бак, где все растворимые материалы конденсируются и удаляются из газа. Перед тем,как попасть в газоочиститель 52, выходящие из камеры догорания газы охлаждаются за счет добавления охлаждающего воздуха 55 или посредством наружной охлаждающей катушки 56. Газы протягиваются через газоочистительную систему вентилятором 57 с диаметром 25 см. 11 Проходящие через вентилятор 57 газы могут быть выброшены в атмосферу. Узел внешнего фильтра 51 и вентилятора предназначен для сбора выбросов загрязнений из тигля. Фильтр 51 имеет два входных шланга,каждый диаметром 10,2 см, которые размещены около крышки тигля. Таким образом, выброшенные загрязнения втягиваются в фильтр 51,фильтруются и выбрасываются в атмосферу. Температура контролировалась в нескольких ключевых точках системы. Температура измерялась двумя термопарами 39, 40, которые вводились через два отдельных отверстия в крышке. Одна термопара 39 измеряла температуру расплава около точки вдувания газа, а вторая термопара 40 использовалась для контроля температуры в верхнем пространстве над расплавом в тигле 44. Примеры и сравнительные испытания Образцы концентрата вольфрама, использованные в этих испытаниях, были получены из коммерческих источников. 817-килограммовый образец был упакован в двух 30-галлонных металлических баках без внутреннего покрытия. Содержимое обоих баков было тщательно перемешано перед началом испытаний. Примерно половина образца была помещена в покрытый пластиком 55-галлоновый бак. Вторая половина образца была разделена на приблизительно 45 килограммовые партии. Из трех партий были взяты пробы для проведения сравнительного анализа, чтобы убедиться в эффективности перемешивания. Дублирующие образцы были оставлены в запасе для проведения контрольных анализов. Перемешанные основные образцы были помещены в герметичные контейнеры до их использования в различных испытаниях. Три пробы концентрата вольфрама были сначала исследованы полуколичественными способами анализа, такими как рентгеновская дифракция, ренттенофлюоресцентная и эмиссионная спектрография, для оценки их минералогического и химического состава. Затем все основные и неосновные компоненты, обнаруженные этими способами, были исследованы более точными способами, включая мокрые количественные способы аналитической химии, спектроскопию атомного поглощения и индуктивную плазменную спектроскопию. В некоторых случаях использовалось несколько способов анализа для точного определения химического состава образца концентрата. Помимо химического анализа, при определении характеристик образца концентрата вольфрама были определены также некоторые физические характеристики. Сравнительные испытания А, В и С В установке с 5-кВт индукционной печью были проведены сравнительные испытания на ошлаковывание, обозначенные А, В и С. С учетом гюбернитовой минералогии образца концентрата, испытания были проведены так, чтобы получить предварительную информацию о 12 свойствах шлака системы MnO-Na2O-SiO2. Компоненты шихты включали в себя различные количества концентрата, хлорида натрия и шлакообразующих веществ - силиката натрия и кремнезема. Они добавлялись в соответствии с относительным составом шихты, подробно описанным в табл. I. Во время проведения экспериментов периодически наблюдались пары избыточной соли и тонкий слой соли, сконденсировавшийся на внутренней поверхности колпака оболочки колпаковой индукционной печи. Таблица I Состав шихты для сравнительных испытаний на ошлаковывание А, В и С Компоненты закладки и масса Концентрат, г Визуальный осмотр полученных в результате шлака и соли показал отсутствие проблем с разделением фаз. Три образца шлака имели стеклообразный вид и были окрашены в зеленый цвет, от светло-зеленого в испытании А до изумрудно-зеленого в испытании С. Большая часть хлорида натрия в первом испытании испарилась, оставив осадок в виде темного твердого вещества на поверхности шлака. В двух других испытаниях галоидная фаза имела не чисто белый цвет и желтый оттенок на поверхности. Образцы шлака и галоидной фазы, полученные в каждом из испытаний, были взяты и подвергнуты рентгенофлюоресцентному анализу; количество галоидной фазы, полученное при проведении испытания А, было недостаточным для проведения рентгенофлюоресцентного анализа. Данные рентгенофлюоресцентного анализа, приведенные в табл. II, показывают, что благоприятное распределение вольфрама между шлаковой и галоидной фазами имело место в двух из трех испытаний. Концентрации оксида вольфрама (WO3) в образцах шлаков, полученных в испытаниях А и С составляли 0,5 и 0,7% соответственно, тогда как концентрация оксида вольфрама в шлаке В составляла 1,8%. Таблица II Результаты рентгенофлюоресцентного анализа образцов шлака, полученных в результате проведения сравнительных испытаний на ошлаковывание Соединение 13 Как видно из табл. III, образцы галоидной фазы испытаний В и С состояли в основном из хлора, натрия и вольфрама. По данным рентгенофлюоресцентного анализа образцы галоидной фазы испытаний В и С содержали 39 и 40% оксида вольфрама соответственно. Концентрации железа и марганца были ниже 0,1% в обоих образцах галоидов. Также были обнаружены микроскопические количества нескольких других элементов. Таблица III Результаты рентгенофлюоресцентного анализа образцов галоидной фазы, полученных в результате предварительных испытаний на ошлаковывание Элемент В вес.% С вес.% Сl 46,5 44,5 Пример 1. Получение вольфрамата натрия. Пример 1 демонстрирует возможность получения отдельной фазы вольфрамата натрия при отсутствии хлорида натрия в шихте. Для успешного проведения испытания необходимо,чтобы вольфрамат натрия и шлак присутствовали как нерастворимые жидкости при интересующей нас температуре. Испытание проводилось в 5-кВт индукционной печи. Состав шихты представлен в табл. IV. Таблица IV Компоненты и массы компонентов шихты для получения вольфрамата натрия Компоненты шихты и их массы Пример 1 Концентрат, г 66,57 Визуальный осмотр полученных в результате испытания продуктов указывает на образование двух отдельных фаз, вольфрамата натрия и шлака. Шлаковая фаза имеет темно-зеленый цвет и находится наверху, а фаза вольфрамата натрия имеет близкий к белому цвет и располагается на дне тигля. То, что фаза вольфрамата натрия находится на дне тигля, связано с ее более высокой плотностью и ее несмешиваемостью с менее плотной шлаковой фазой. Проведение испытания примера 1 не вызвало трудностей и было достигнуто отличное разделение фаз. Полученные в результате эксперимента продукты были подвергнуты анализу с целью определения относительного распределения вольфрама и марганца между двумя фазами. Как показано в табл. V, результаты анализов примера 1 подтвердили возможность получения благоприятного распределения вольфрама и марганца между шлаковой и соляной фазами. Судя по этим результатам, выделение вольфрама из концентрата в виде вольфрамата натрия 14 при отсутствии хлорида натрия представляет собой многообещающую альтернативу. Таблица V Результаты анализов примера 1 Состав шихты Результаты химического анализа Вещества Вес.% Элемент % в соли % в шлаке Сравнительные испытания D, E, F и G Изменение отношения содержания марганца к содержанию силиката натрия в шихте Целью проведения сравнительного испытания D было определить влияние большого отношения содержания марганца к содержанию силиката натрия (Mn:Na2SiO3) на распределение вольфрама и марганца между шлаковой и соляной фазами. Не желая ограничивать себя рамками какой-либо теории, мы предполагаем, что важно поддерживать определенный уровень содержания натрия в системе. При проведении испытаний, в которых состав шлака являлся независимой переменной, относительное распределение марганца между соляной и шлаковой фазами ухудшалось при повышении содержания оксида марганца в шлаке. Эта проблема стала очевидной, когда молярное отношение содержания марганца к содержанию силиката натрия в шихте было повышено до значения,превосходящего 1. Считается, что это явление связано с недостаточным количеством натрия для осуществления следующей реакции:MnWO4(s)+Na2SiO3(s)=MnOSiO2(1)+Na2WO4(1) Если желательно иметь отношение содержания Mn:Na2SiO3, превосходящие единицу, для уменьшения расхода плавня, то необходим дополнительный источник натрия для максимального повышения образования вольфрамата натрия и минимизации растворения непрореагировавшего вольфрамата марганца в соляной фазе. Дополнительным источником натрия, использованным в этих экспериментах, служил гидроксид натрия (NaOH) в соответствии с составом, представленным в табл. VI. Таблица VI Состав шихты для испытания D Соединение Испытание D Концентрат, г 41,23 Хотя в этом случае также образовывались две отдельные фазы, внешний вид шлака отличался от того, который наблюдался в предыдущих экспериментах. Полученный в результате эксперимента шлак был похож на песок, наблюдение которого под микроскопом (30-кратное увеличение) выявило наличие различных фаз и В испытании F была сделана попытка увеличить отношение содержания марганец:кремний до 1,35 при условиях, близких к условиям испытания D, но при использовании карбоната натрия (Na2CO3) вместо гидроксида натрия в качестве дополнительного источника натрия. Была поставлена цель определить, влияет ли выбор дополнительного источника натрия на распределение вольфрама и марганца. Как видно из табл. VII, концентрация вольфрама в шлаке 1,08% по сравнению с концентрацией 3,9%, полученной в испытании D, позволяет предположить, что дополнительный источник натрия оказывает влияние на распределение вольфрама в галоидной фазе. Целью испытания G было изучение возможности использования кремнезема и карбоната натрия в качестве единственных источников натрия и кремнезема как попытки заменить метасиликат натрия на более дешевое сырье. Как видно из табл. VII, концентрация вольфрама в шлаке 0,13% и концентрация марганца в галоиде 0,03% позволяет предположить, что такая замена возможна. Таблица VII Результаты анализов сравнительных испытаний D, Е, F и G Состав шихты Результаты химического анализа Комментарии Вещество Вес.% ЭлементW 17,50 9,44 качестве дополнительного источника натрия дляW 23,40 1,08 качестве дополнительного источника натрия (основNaCl 50,28W 23,60 0,13 лись в качестве единственных источников кремNaCl 37,36 неполное расплавление шихты. Галоидная фаза имела такой же вид, как и в предыдущих экспериментах. Как видно из результатов анализов,приведенных в табл. VII, использование гидроксида натрия дает неудовлетворительное распределение вольфрама и марганца в шлаковой и соляной фазах. Для дальнейшего изучения влияния изменения отношения содержания Mn:Na2SiO3 в шихте, а также для изучения возможности использования альтернативных источников натрия и кремнезема, были проведены три дополнительных испытания в индукционной печи. Целью испытания Е было изучение влияния состава шлака с молярным отношением Mn:Na2SiO3,равным единице, на распределение вольфрама и марганца между галоидной и соляной фазами. Как видно из табл. VII, концентрация вольфрама в шлаке 0,55% и концентрация марганца в галоиде 0,14% указывает на то, что уменьшение расхода плавня для достижения молярного отношения Mn:Na2SiO3, меньшего или равного единице, возможно и заслуживает внимания. Пример 2. В примере 2 хлорид натрия был включен в состав шихты. Состав шлака выбирался так,чтобы обеспечить образование несмешиваемых жидкостей. Образование двух несмешиваемых фаз, также как и благоприятное распределение вольфрама и марганца между солью и шлаком,были предсказаны по результатам примера 1. Массовый баланс примера 2 приведен в табл. VIII. Итоговая масса в этом примере составила 96,8%. Массовый баланс вольфрама указывает на то, что 92,9% вольфрама шихты сконцентрировалось в галоидной фазе, и 3,3%17 в шлаке. Только 3,8% вольфрама шихты остались неучтенными. Таблица VIII Массовый баланс процесса ошлаковывания примера 2 Вещество Вещество Килограммы Килограммы на входе на выходе Концентрат 4,43 Галоидная фаза 3,94 Кремнезем 1,41 Шлак 5,74 Метасиликат натрия 4,15 Всего 10,00 Всего 9,68 Распределение вольфрама для примера 2,рассчитанное по результатам анализа, указывает на то, что приблизительно 97% исходного вольфрама концентрируется в вольфраматовой фазе, остальная часть - в шлаке. Хотя эти результаты позволяют предположить, что эффективное ошлаковывание при отсутствии хлорида натрия возможно, специалисты в данной области могут улучшить распределение вольфрама без проведения лишних экспериментов. Захват соли со стенок реактора во время сливания шлака может вносить свой вклад в высокую концентрацию вольфрама в шлаке в примере 2. Из технологических соображений, хотя фаза вольфрамата натрия выделяется в реакторе под шлаком (из-за более высокой плотности соли), она сливается первой, что связано с ее меньшей вязкостью и геометрией реактора. В соли обнаружены только минимальные количества захваченного шлака, которые плавают на поверхности до затвердевания соли в ковше. Пример 3. Повторное использование прошедшей барботирование отработанной соли в операции ошлаковывания. Этот пример иллюстрирует эффективность повторного использования отработанной соли из барботажного устройства в операции ошлаковывания. Такая практика является желательной по двум причинам. Во-первых, отработанная соль обычно содержит значительную концентрацию вольфрама (примерно 15 вес.%), не превращенного в карбид вольфрама во время барботирования. Повторное использование соли в барботировании позволяет сохранить вольфрам в цикле переработки и достичь высокоэкономичного уровня извлечения вольфрама во всей операции переработки. Во-вторых, счита 001768 18 ется, что оксид натрия, содержащийся в прошедшей барботирование соли, может служить источником натрия при получении вольфрамата натрия во время барботирования. Поэтому ожидается, что повторное использование отработанной соли уменьшит расход сырья. Эксперимент этого примера был проведен для того, чтобы определить, повлияет ли повторное использование отработанной соли на распределение вольфрама между соляной и шлаковой фазами. Эксперимент этого примера состоит из трех последовательных циклов закладки шихты. Шихта цикла 1 - это типичная шихта для ошлаковывания без повторного использования соли,прошедшей барботирование, и представляет собой 5 кг смеси 26,3% концентрата гюбернита,24,7% метасиликата натрия (Na2SiO3), 8,4% кремнезема (SiO2) и 40,6% хлорида натрия(NaCl). Шихта цикла 2 состоит из 1 кг отработанной соли (полученной во время предыдущего испытания на барботирование) плюс 4 кг тех же самых компонентов, которые были использованы в цикле 1, смешанных в таких же пропорциях. Шихта цикла 3 содержит 2 кг отработанной соли плюс 4 кг компонентов, использованных в цикле 1 и смешанных в пропорциях,использованных в цикле 1. В начале каждого цикла материалы шихты смешиваются вместе, закладываются в печь и обрабатываются при номинальной температуре 1050 С в течение одного часа. Затем удаляется расплавленная соль, для чего печь наклоняется и соль сливается в ковш, где она затвердевает. Печь возвращается в рабочее вертикальное положение и процесс повторяется со следующей шихтой. Шлаковая фаза не удаляется, пока не будут завершены все три цикла. Затем шлак сливается в ковш и застывает. Были приготовлены и подвергнуты химическому анализу образцы шлака и всех солей, полученных во всех трех циклах. Результаты анализа шлака и соли приведены в табл. IX. Как было рассчитано на основании измеренных весов продуктов и анализа содержания вольфрама, приблизительно 99,1% вольфрама остается в соли. Сравнительный анализ соли, полученной в циклах 1, 2 и 3,показывает достаточно низкие концентрации примесей для всех трех циклов. Таблица IX Результаты анализа испытаний на повторное использование отработанной соли Пример 3. ОпиМасса Аl% Сl% Сr%W% сание образцов образца, кг Соль цикла 1 3,01 0,01 36 0,002 0,01 0,09 29,4 0,007 0,1 28,4 Соль цикла 2 3,57 0,01 40 0,003 0,01 0,04 31,5 0,002 0,1 21,9 Соль цикла 3 4,62 0,01 41 0,002 0,01 0,04 31,6 0,002 0,1 22,8 Шлак 4,21 0,09 2,5 0,08 0,34 13,9 17,1 0,13 24,8 0,6 Примеры 4 и 5. Барботирование вольфрамата натрия для получения карбида натрия. Было показано, что можно получить карбид вольфрама довольно высокой чистоты путем барботирования расплавленной смеси 19 вольфрамата натрия и хлорида натрия метаном. Однако исключение хлорида натрия из процесса улучшает способ по нескольким причинам. Чтобы продемонстрировать, что порошок кристаллического карбида вольфрама можно получить путем барботирования расплавленного вольфрамата натрия метаном, приведены два следующих примера. В обоих примерах исходный расплав состоит из вольфрамата натрия, полученного в результате проведения предыдущих испытаний на шлакообразование. Во время проведения экспериментов этих примеров ванна жидкого вольфрамата натрия поддерживалась при температуре приблизительно 1100 С, а газообразный метан нагнетался под поверхность расплава со скоростью приблизительно 11,4 л/мин. Барботирование метаном продолжалось в течение 3 ч в примере 4 и в течение 90 мин в примере 5. По завершении выполнения экспериментов обоих примеров расплавленные продукты сливались в стальной ковш и затвердевали. После затвердения и достаточного охлаждения продуктов наблюдались две отдельные фазы: белый отработанный пепел и более плотная серая фаза. Эти две фазы были разделены и подготовлены для проведения рентгеновского дифракционного анализа. Для рентгеновского дифракционного анализа были выбраны продукты эксперимента примера 4, так как они были чище и легче разделялись после охлаждения. Рентгеновский дифракционный анализ белой отработанной соли показал, что она в основном состоит из непрореагировавшего вольфрамата натрия (Na2WO4) и содержит микроконцентрации других неотождествленных кристаллических фаз. Что более важно, было обнаружено, что плотная фаза содержит вольфрамат натрия при меньших концентрациях карбида вольфрама WC и карбида вольфрама W2C при микроскопических концентрациях металлического вольфрама и той же самой неотождествленной кристаллической фазы, что и в белой соли. Хотя опытные специалисты в данной области смогут оптимизировать этот процесс, результаты рентгеновской дифракции показали,что возможно получать карбид вольфрама способом барботирования метаном. Хотя здесь подробно описаны различные варианты осуществления настоящего изобретения, очевидно, что специалисты смогут внести изменения и улучшения в эти варианты осуществления изобретения. Следует ясно понимать,что такие изменения и улучшения не выходят за рамки сущности настоящего изобретения, описываемого приведенной ниже формулой. Ссылочная нумерация Вольфрамсодержащий концентрат 1 Кремнезем 2 Силикат 3 Ошлаковывающая печь 4 20 Вольфрамат 5 Шлак 6 Барботажная печь 7 Газ из ошлаковывающей печи 8 Контроль на содержание твердых частиц 9 Отработанный газ Углеродсодержащий газ 10 Газ из барботажной печи 11 Камера догорания 12 Кислородсодержащий газ 13 Метан 14 Газ из камеры догорания Контроль на содержание твердых частиц 15 Твердые частицы Отработанный газ 16 Прошедшая барботирование отработанная соль 17 Неочищенный карбид вольфрама 18 Этап водного выщелачивания 19 Вода 20 Разделение твердых веществ и жидкости 21 Жидкая составляющая 22 Кристаллизатор 23 Кристаллы 24 Кристаллы твердого неочищенного карбида вольфрама 25 Дробление 26 Вода 27 Кислотное выщелачивание 28 Кислота 29 Прошедшая дробление и кислотное выщелачивание суспензия 30 Разделение твердых веществ и жидкости 31 Высокочистый карбид вольфрама 32 Раствор 33 Нейтрализации и осаждение 34 Твердый осадок 35 Наклоняющаяся печь 36 Трубки для барботирования 37, 38 Термопары 39, 40 Труба для подачи азота 41 Линия вывода 42 Манометр 43 Тигель 44 Азот 45 Газообразный углеводород(ы) 46 Расходомеры 47, 48, 49 Камера догорания 50 Фильтр 51 Газоочиститель 52 Кислород 53 Клапан 54 Забор охлаждающего воздуха 55 Охлаждающая катушка 56 Вентилятор 57 21 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ концентрирования вольфрама из вольфрамсодержащего материала, включающий следующие этапы:(a) нагрев вольфрамсодержащего материала в присутствии неорганического соединения щелочного металла и при практически полном отсутствии галоидной соли при температуре,достаточной для расплавления упомянутого вольфрамсодержащего материала в течение времени, достаточного для образования фазы с большей плотностью и фазы с меньшей плотностью, причем большая часть упомянутого вольфрама концентрируется в фазе с большей плотностью в виде соли вольфрама,(b) разделение упомянутой фазы с большей плотностью и упомянутой фазы с меньшей плотностью под действием силы тяжести, причем упомянутая фаза с большей плотностью оседает на дно,(c) концентрирование упомянутого вольфрама путем отделения упомянутой фазы с большей плотностью от фазы с меньшей плотностью,(d) получение карбида вольфрама путем барботирования упомянутой фазы с большей плотностью углеродсодержащим газом при повышенной температуре с целью получения карбида вольфрама, и(e) рециклирование части солесодержащего материала из барботированной фазы с большей плотностью этапа (d) в расплав, содержащий вольфрамсодержащий материал, в присутствии соединения щелочного металла, упомянутого выше в этапе (а). 2. Способ по п.1, в котором упомянутый вольфрамсодержащий материал выбирают из группы, состоящей из гюбнерита (MnWO4),шеелита (CaWO4), ферберита (FeWO4), вольфрамита Fe,Mn)WO4) и их смесей. 3. Способ по п.1, в котором упомянутый вольфрамсодержащий материал выбирают из группы, состоящей из колошниковой пыли,шлаков, лома и их смесей. 4. Способ по п.1, в котором упомянутое соединение щелочного металла выбирают из группы, состоящей из соединений натрия и соединений калия. 5. Способ по п.1, в котором упомянутое соединение щелочного металла выбирают из группы, состоящей из силиката натрия, карбоната натрия, гидроксида натрия и их смесей. 6. Способ по п.1, в котором этап нагревания (а) производят при температуре от приблизительно 900 С до приблизительно 1200 С. 7. Способ по п.1, в котором этап нагревания (а) производят при температуре от приблизительно 1050 С до приблизительно 1150 С. 8. Способ по п.1, в котором этап барботирования (d) производят при температуре от приблизительно 1050 С до приблизительно 1200 С. 22 9. Способ по п.1, в котором этап барботирования (d) производят при температуре от приблизительно 1050 С до приблизительно 1150 С. 10. Способ по п.1, в котором упомянутым углеродсодержащим газом является газообразный углеводород. 11. Способ по п.1, в котором упомянутый углеродсодержащий газ выбирают из группы,состоящей из метана, этилена, пропана и их смесей. 12. Способ по п.1, в котором упомянутую фазу с высокой плотностью отделяют от упомянутой фазы с низкой плотностью путем сливания упомянутой фазы с высокой плотностью из тигля. 13. Способ по п.1, в котором упомянутую фазу с высокой плотностью удаляют через отверстие в тигле, содержащем упомянутую фазу с высокой плотностью и упомянутую фазу с низкой плотностью. 14. Способ по п.1, в котором упомянутый карбид вольфрама, образованный на этапе (d),отделяют от остального материала и очищают. 15. Способ по п.14, в котором упомянутый карбид вольфрама имеет чистоту не менее 90% после очистки. 16. Способ получения карбида вольфрама,включающий следующие этапы:(a) нагрев концентрата вольфрамсодержащего минерала в присутствии неорганического соединения натрия или калия при практически полном отсутствии хлорида натрия до температуры от приблизительно 900 С до приблизительно 1200 С для получения первичного расплава,(b) поддержание упомянутого первичного расплава при температуре от приблизительно 900 С до приблизительно 1200 С до тех пор,пока упомянутый расплав не разделится на вольфрамсодержащую фазу с большей плотностью и шлаковую фазу с меньшей плотностью,причем, по меньшей мере, 80% вольфрама в упомянутом вольфрамовом минеральном концентрате концентрируется в упомянутой вольфрамсодержащей фазе с большей плотностью,(c) отделение упомянутой вольфрамсодержащей фазы с большей плотностью от шлаковой фазы с меньшей плотностью,(d) нагрев упомянутой вольфрамсодержащей фазы с большей плотностью до температуры от приблизительно 1050 С до приблизительно 1200 С для получения вторичного расплава,(e) пропускание газообразного метана через упомянутый вторичный расплав для получения барботированного вторичного расплава,содержащего карбид вольфрама,(f) отделение части расплава, обогащенной карбидом вольфрама, от упомянутого барботированного вторичного расплава, и(g) очистка упомянутой части расплава,обогащенной карбидом вольфрама, для получения очищенного карбида вольфрама. 23 17. Способ по п.16, в котором часть солесодержащего материала берут из упомянутого вторичного расплава и помещают (рециклируют) в первичный расплав типа упомянутого в этапе (а) для участия в отделении упомянутой вольфрамсодержащей фазы с большей плотностью от шлаковой фазы с меньшей плотностью. 18. Способ по п.16, в котором упомянутая вольфрамсодержащая фаза с большей плотностью содержит соль оксида вольфрама. 19. Способ по п.16, в котором упомянутая вольфрамсодержащая фаза с большей плотностью содержит соль вольфрамата натрия. 20. Способ по п.16, в котором упомянутая шлаковая фаза с меньшей плотностью содержит силикат. 21. Способ по п.16, в котором упомянутая шлаковая фаза с меньшей плотностью содержит силикат марганца, силикат железа или силикаты алюминия-кальция. 22. Способ по п.16, в котором упомянутый карбид вольфрама образуется в виде мелких частиц, причем не менее 90% частиц карбида вольфрама имеют средний диаметр менее 10 мкм. 23. Способ по п.16, в котором не менее 95% вольфрама в упомянутом вольфрамсодержащем минерале концентрируется в упомянутой вольфрамсодержащей фазе с большей плотностью. 24. Способ по п.16, в котором не менее 97% вольфрама в упомянутом вольфрамсодержащем минерале концентрируется в упомянутой вольфрамсодержащей фазе с большей плотностью. 25. Способ по п.16, в котором, по крайней мере, часть упомянутого соединения натрия выбирают из группы, состоящей из: (i) силиката натрия (Nа 2SiO3) или (ii) карбоната (Nа 2 СО 3) в присутствии кремнезема (SiO2). 26. Способ по п.16, в котором упомянутый этап очистки (g) включает:(a) сухое дробление, воздушную сепарацию и сухую сепарацию упомянутой части расплава, обогащенной карбидом вольфрама,(b) водное выщелачивание упомянутого очищенного сухим способом материала,(c) разделение прошедшего водное выщелачивание материала на твердую и жидкую фазы для получения кристаллов карбида вольфрама,(d) дробление и кислотное выщелачивание упомянутых кристаллов неочищенного карбида вольфрама, и(e) разделение прошедшего дробление и кислотное выщелачивание кристаллического неочищенного карбида вольфрама на твердую и жидкую фазы для получения высокочистого карбида вольфрама. 27. Способ по п.16, в котором упомянутый очищенный карбид вольфрама имеет содержание карбида вольфрама не менее 90%. 24 28. Способ получения карбида вольфрама,включающий следующие этапы:(а) нагрев соли вольфрамовой кислоты до температуры, превосходящей температуру ее плавления, для получения расплава при практически полном отсутствии каких-либо галоидных солей, разделение расплава на фазу с большей плотностью и фазу с меньшей плотностью, причем большая часть вольфрама концентрируется в упомянутой фазе с большей плотностью,(b) пропускание газообразного углеводорода через упомянутую фазу с большей плотностью для получения карбида вольфрама, и(c) отделение упомянутого карбида вольфрама от остальной фазы с большей плотностью. 29. Способ по п.28, в котором упомянутой солью вольфрамовой кислоты является вольфрамат натрия. 30. Способ получения карбида вольфрама,включающий следующие этапы:(a) нагрев концентрата вольфрамсодержащего минерала в присутствии неорганического соединения натрия или калия при практически полном отсутствии хлорида натрия до температуры от приблизительно 900 С до приблизительно 1200 С для получения первичного расплава,(b) поддержание упомянутого первичного расплава при температуре от приблизительно 900 С до приблизительно 1200 С до тех пор,пока упомянутый расплав не разделится на вольфрамсодержащую фазу с большей плотностью и шлаковую фазу с меньшей плотностью,(c) отделение упомянутой вольфрамсодержащей фазы с большей плотностью от шлаковой фазы с меньшей плотностью,(d) нагрев упомянутой вольфрамсодержащей фазы с большей плотностью до температуры от приблизительно 1050 С до приблизительно 1200 С для получения вторичного расплава,(e) пропускание газообразного метана через упомянутый вторичный расплав для получения барботированного вторичного расплава,содержащего карбид вольфрама,(f) отделение части расплава, обогащенной карбидом вольфрама, от упомянутого барботированного вторичного расплава, и(g) очистка упомянутой части расплава,обогащенной карбидом вольфрама, для получения очищенного карбида вольфрама. 31. Способ по п.5, в котором упомянутый солесодержащий материал, подвергаемый рециклированию, представляет собой материал,выбранный из группы, состоящей из вольфрамата натрия, Na2O и их смесей. 32. Способ по п.17, в котором упомянутый рециклируемый солесодержащий материал представляет собой материал, выбранный из группы, состоящей из вольфрамата натрия,Na2O и их смесей. 33. Способ по п.29, в котором часть упомянутой оставшейся фазы с большей плотно 25 стью этапа (е) рециклируют в расплав типа упомянутого в этапе (а). 34. Способ по п.26, в котором жидкую фазу, отделяемую на этапе (с), подают в кристал Фиг. 1
МПК / Метки
МПК: C01B 31/30
Метки: обработки, способ, пирометаллургический, металлсодержащих, материалов
Код ссылки
<a href="https://eas.patents.su/15-1768-pirometallurgicheskijj-sposob-obrabotki-metallsoderzhashhih-materialov.html" rel="bookmark" title="База патентов Евразийского Союза">Пирометаллургический способ обработки металлсодержащих материалов</a>
Устройство для обработки упаковочных материалов и способ изготовления упаковочных контейнеров

Номер патента: 1122
Опубликовано: 30.10.2000
Авторы: Кацумата Сигео, Морияма Ясуюки, Кумета Юкихиса
Метки: материалов, контейнеров, упаковочных, изготовления, устройство, способ, обработки
Формула / Реферат:
1. Устройство для обработки упаковочного материала, содержащее (a) транспортирующее средство для перемещения многослойного упаковочного материала, который образован из бумажной основы и полимерных пленок и который имеет зону разрыва в месте, соответствующем положению открывающего средства; (b) запечатывающее устройство для запечатывания указанного упаковочного материала; и (c) нагревательное средство, расположенное в направлении перемещения...
Способ разделения смешанных порошковых материалов

Номер патента: 326
Опубликовано: 29.04.1999
Автор: Нельсон Бенджамин В.
МПК: B04B 1/00
Метки: смешанных, порошковых, материалов, разделения, способ
Формула / Реферат:
1. Способ разделения смешанных порошковых материалов разного удельного веса в суспензии, включающий в себя вращение барабана центрифуги, имеющего периферийную стенку и открытую входную часть, вокруг продольной оси совместно с периферийной стенкой, загрузку материалов в барабан с обеспечением дальнейшего их прохождения по периферийной стенке и побуждение более тяжелых частиц улавливаться на периферийной стенке, а более легких частиц выходить в...
Способ ломки материалов на заготовки и устройство для его осуществления.

Номер патента: 265
Опубликовано: 25.02.1999
Автор: Жалдак Николай Иванович
МПК: B23D 27/06
Метки: осуществления, устройство, способ, материалов, ломки, заготовки
Формула / Реферат:
1. Способ ломки материалов на заготовки, включающий нанесение на материал концентратора напряжений в виде углублений и приложение в его плоскости разрушающей импульсной нагрузки, отличающийся тем, что материал по границе углубления концентратора напряжения сначала сжимают усилием до предела упругости, а в зоне образования углубления концентратора напряжений - до предела пластичности и образования большого волнового сопротивления в пределах...
Способ биообработки твердых материалов в поверхностном биореакторе без перемешивания

Номер патента: 429
Опубликовано: 24.06.1999
Автор: Кор Уильям Дж.
МПК: B01D 11/00, C22B 1/00, C01G 7/00...
Метки: биореакторе, перемешивания, биообработки, твердых, поверхностном, материалов, способ
Формула / Реферат:
1. Способ биообработки твердого материала для удаления нежелательного соединения с применением поверхностного биореактора без перемешивания, включающий стадии: а.) покрытия поверхности множества крупнозернистых подложек, имеющих размер частиц более чем 0,3 см, подвергаемым биообработке твердым материалом и образования вследствие этого множества покрытых крупнозернистых подложек, при этом биообрабатываемый твердый материал имеет размер частиц...
Способ манипуляции изображениями напольных покрытий или других материалов и система для его осуществления.

Номер патента: 989
Опубликовано: 28.08.2000
Авторы: Томас Гленн С., Ван Варк Джей
МПК: G06T 11/40
Метки: манипуляции, система, осуществления, других, напольных, способ, изображениями, покрытий, материалов
Формула / Реферат:
1. Способ манипуляции с помощью компьютера характеристиками многоцветных изображений в цифровой форме материалов, в котором материал содержит, по крайней мере, одну позицию, причем каждая позиция содержит подмножество в материале, которому должна быть придана в существенной степени та же самая обработка, и в котором изображение материала сохранялось в памяти компьютера в считываемом и манипулируемом формате, и одна или более позиций материала...
Предыдущий патент: Способ обработки дизельного топлива
Следующий патент: Антагонисты рецепторов возбудительной аминокислоты
Случайный патент: Способ получения ингибитора протеазы вич из(2-s)-4-пиколил-2-пиперазин-трет-бутилкарбоксамида