Гликозидирование индолкарбазола с применением межфазного катализа
Номер патента: 5209
Опубликовано: 30.12.2004
Авторы: Сатаке Нобуя, Хирага Соуити, Россен Кай, Вайссман Стивен А., Петрилло Дэниел Е.








Формула / Реферат
1. Способ получения соединения формулы I

в которой Q является O, N-R, S или CH2;
X1 и X2 независимо выбираются из
1) H,
2) галогена,
3) OH,
4) CN,
5) NC,
6) CF3,
7) (C=O)NO2,
8) (C=O)C1-C6алкила,
9) (C=O)OC1-C6алкила,
10) OCH2OCH2CH2Si(CH3)3,
11) NO2,
12) 9-флуоренилметилкарбонила,
13) NR5R6,
14) OC1-C6алкила,
15) C1-C6алкила,
16) C1-C6алкиленарила и
17) OC1-C6алкиленарила;
R и R1 являются независимо
1) H,
2) (C=O)C1-C6алкилом,
3) (C=O)CF3,
4) (C=O)OC1-C6алкилом,
5) 9-флуоренилметилкарбонилом,
6) группой фуранозы или
7) группой пиранозы,
пока при условии, что один из R и R1 является группой фуранозы или группой пиранозы;
R2 и R3 являются независимо OH или H, или R2 и R3 вместе обозначают оксогруппу;
R4 является
1) H,
2) C1-C10алкилом,
3) CHO,
4) (C=O)C1-C10алкилом,
5) (C=O)OC1-C10алкилом,
6) C0-C10алкиленарилом или
7) C0-C10алкилен-NR5R6;
R5 и R6 независимо являются
1) H,
2) (C1-C8алкил)-(R7)2,
3) (C=O)O(C1-C8алкилом),
4) 9-флуоренилметилкарбонилом,
5) OCH2OCH2CH2Si(CH3)3,
6) (C=O)(C1-C8алкилом),
7) (C=O)CF3 или
8) (C2-C8алкенил)-(R7)2 или
R5 и R6 совместно с азотом, к которому они присоединены, образуют N-фталимидогруппу;
R7 является
1) H,
2) OH,
3) OC1-C6алкилом или
4) арилом, причем указанный арил необязательно замещен, с числом замещающих групп до двух, выбранных из OH, O(C1-C6алкила) и (C1-C3алкилен)-OH;
который включает следующие стадии:
(а) взаимодействие фуранозы или пиранозы с активирующим реагентом для получения активированного сахара и
(б) присоединение активированного сахара к соединению формулы IV

где Rla является H, если Q является O, S, CH2 или N-R и R не является H, иначе Rla выбирают из R1;
в присутствии водного раствора гидроксида щелочного металла и межфазного катализатора в двухфазной системе с получением соединения формулы I.
2. Способ по п.1, в котором R и R1 независимо выбираются из группы фуранозы формулы IIA или группы пиранозы формулы IIB, когда R или R1 определены как группа фуранозы или группа пиранозы соответственно

R8 независимо выбирается из
1) водорода,
2) C1-C6алкила,
3) OH,
4) галогена,
5) O(C1-C6алкила),
6) O(C1-C6алкилен)арила,
7) OSO2(C1-C6алкила),
8) OSO2арила,
9) OCH2OCH2CH2Si(CH3)3,
10) O(C=O)(C1-C6алкила),
11) O(C=O)CF3,
12) азидо или
13) NR5R6 или
два R8 на одном атоме углерода вместе образуют оксогруппу, =N-R5 или =N-R7; и
фураноза или пираноза на стадии (а) являются фуранозой формулы IIIA или пиранозой формулы IIIB соответственно

3. Способ по п.2, в котором активирующий реагент на стадии (a) выбирают из галогенангидрида, и двухфазная система на стадии (b) включает органический растворитель, выбранный из углеводорода, нитрила, простого эфира, галогенированного углеводорода, кетона или неполярного апротонного растворителя.
4. Способ по п.3, в котором активирующий реагент выбирают из SOCl2 или оксалилхлорида.
5. Способ по п.3, в котором двухфазная система составляется из метил-трет-бутилового эфира, дихлорметана или трифтортолуола.
6. Способ по п.3, в котором межфазным катализатором на стадии (б) является (Ra)4M+A-;
Ra независимо является H или C1-C18алифатическим углеводородом;
M является N или P; и
A является OH, F, Br, Cl, I, HSO4, CN, MeSO3 или PhCH2CO2.
7. Способ по п.6, в котором межфазным катализатором является трикаприлметиламмоний хлорид.
8. Способ по п.3, в котором водный раствор гидроксида щелочного металла на стадии (б) имеет концентрацию от примерно 5 до примерно 95% мас./мас. и гидроксид щелочного металла выбран из гидроксида лития, гидроксида натрия, гидроксида калия и гидроксида цезия.
9. Способ по п.8, в котором водный раствор гидроксида щелочного металла имеет концентрацию от примерно 45 до примерно 50% мас./об. и гидроксид щелочного металла является гидроксидом калия или гидроксидом натрия.
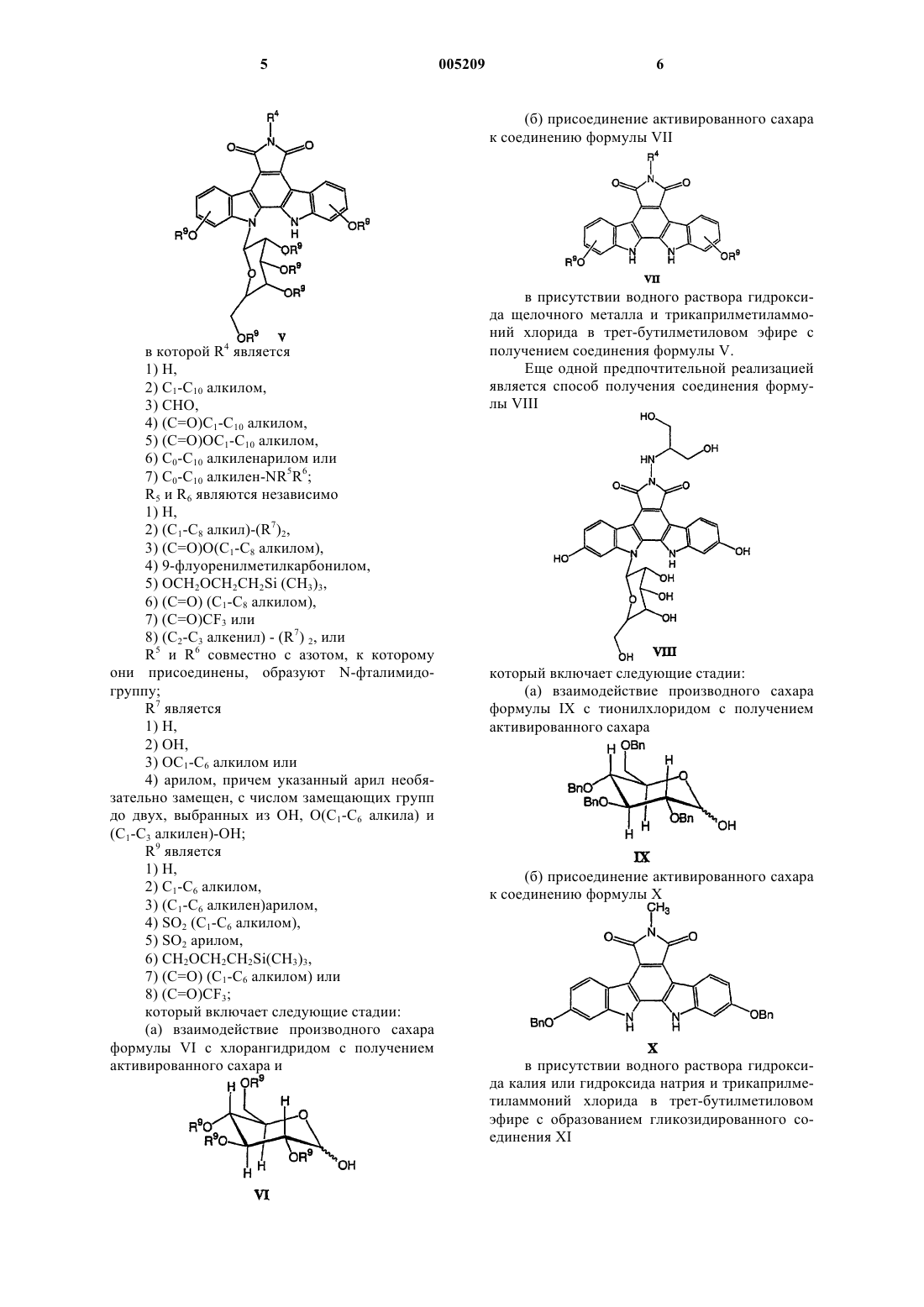
10. Способ получения соединения формулы V

в котором R4 является
1) H,
2) C1-C10алкилом,
3) CHO,
4) (C=O)C1-C10алкилом,
5) (C=O)OC1-C10алкилом,
6) C0-C10алкиленарилом или
7) C0-C10алкилен-NR5R6;
R5 и R6 являются независимо
1) H,
2) (C1-C8алкил)-(R7)2,
3) (C=O)O(C1-C8алкилом),
4) 9-флуоренилметилкарбонилом,
5) OCH2OCH2CH2Si(CH3)3,
6) (C=O)(C1-C8алкилом),
7) (C=O)CF3 или
8) (C2-C8алкенил)-(R7)2, или
R5 и R6 совместно с азотом, к которому они присоединены, образуют N-фталимидогруппу;
R7 является
1) H,
2) OH,
3) OC1-C6алкилом или
4) арилом, причем указанный арил необязательно замещен, с числом замещающих групп до двух выбранных из OH, O(C1-C6алкила) и (C1-C3алкилен)-OH;
R9 является
1) H,
2) C1-C6алкилом,
3) (C1-C6алкилен)арилом,
4) SO2(C1-C6алкилом),
5) SO2арил,
6) CH2OCH2CH2Si(CH3)3,
7) (C=O)(C1-C6алкилом) или
8) (C=O)CF3;
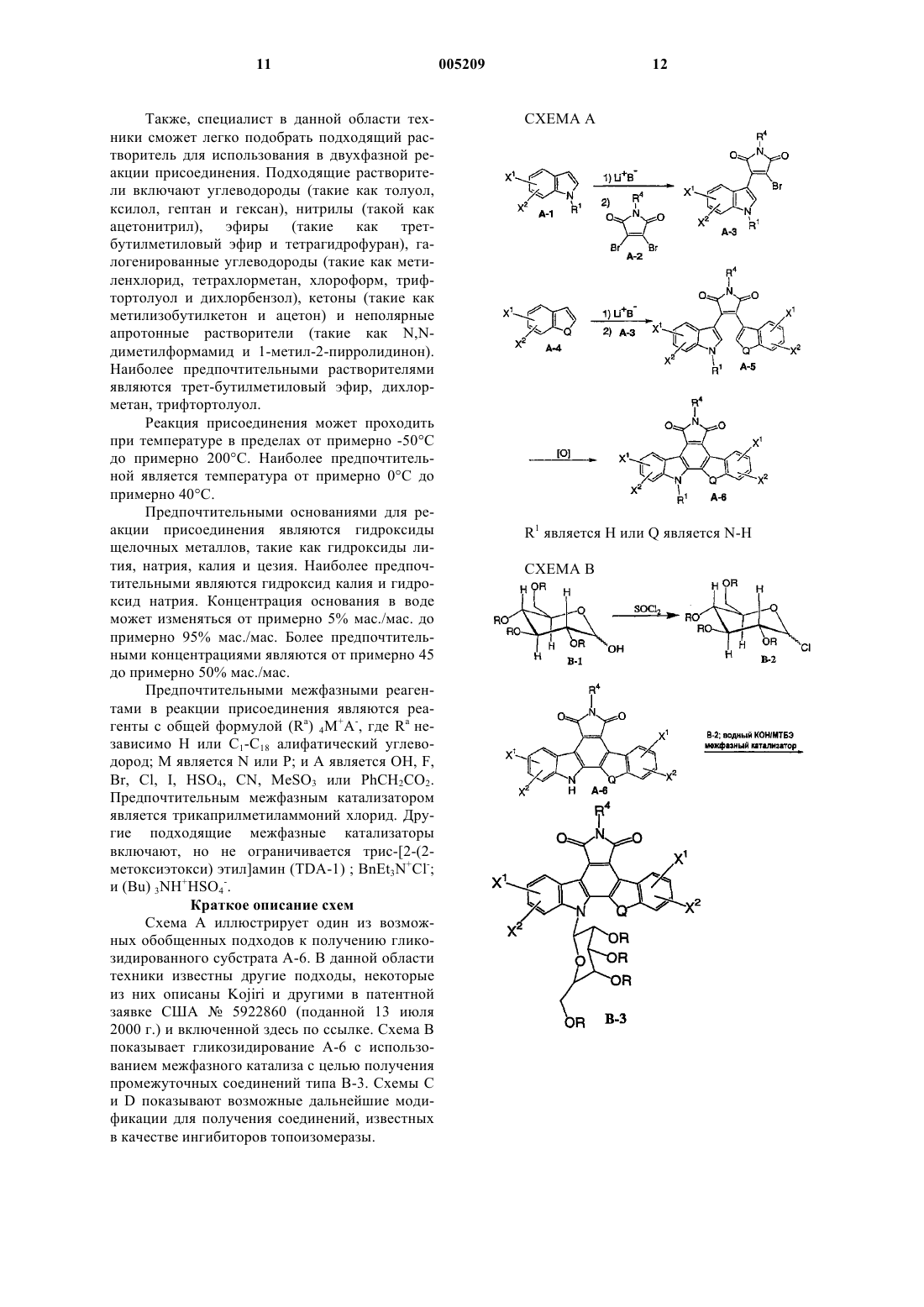
который включает следующие стадии:
(а) взаимодействие производного сахара формулы VI с хлорангидридом с получением активированного сахара и

(б) присоединение активированного сахара к соединению формулы VII

в присутствии водного раствора гидроксида щелочного металла и трикаприлметиламмоний хлорида в трет-бутилметиловом эфире с получением соединения формулы V.
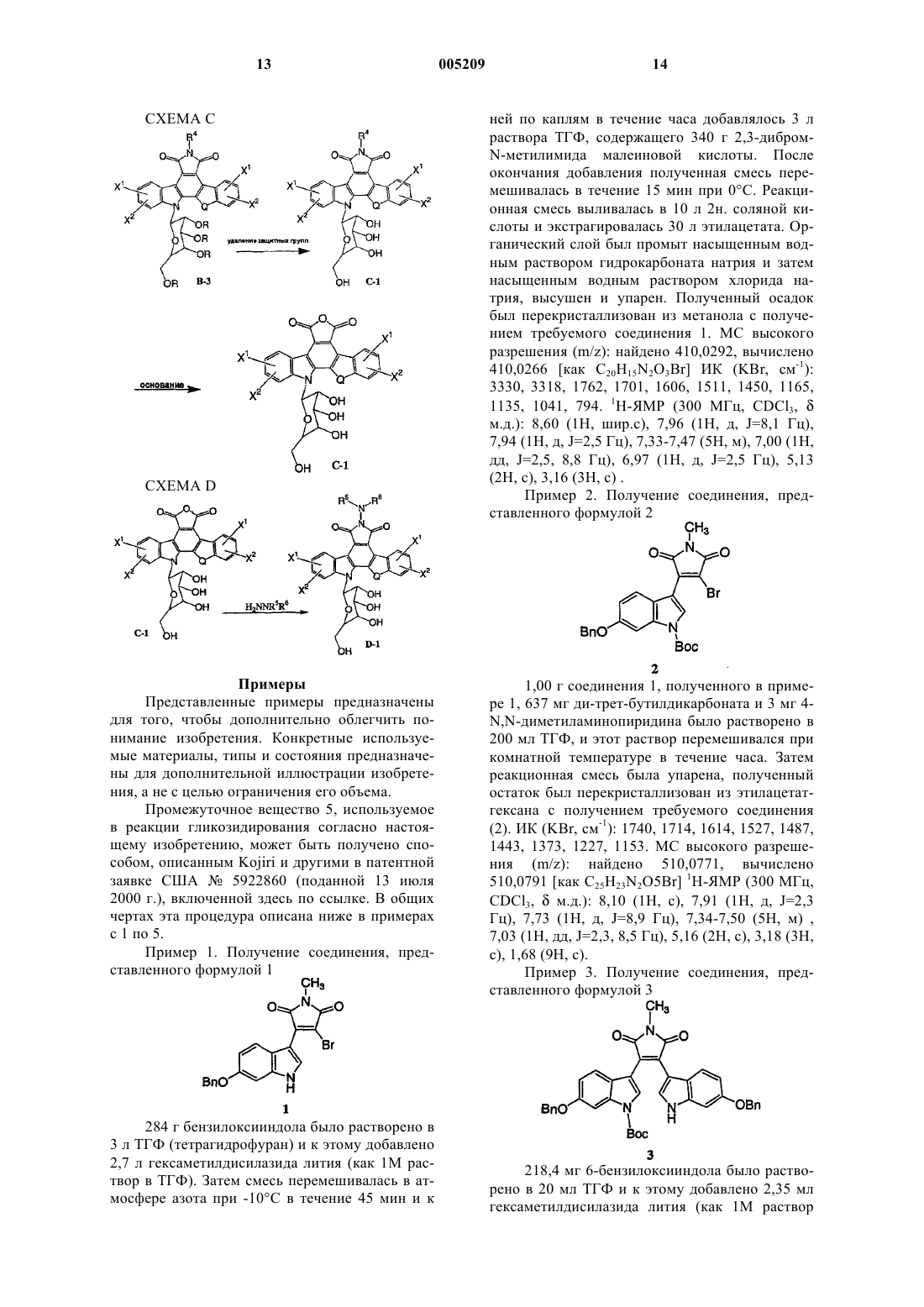
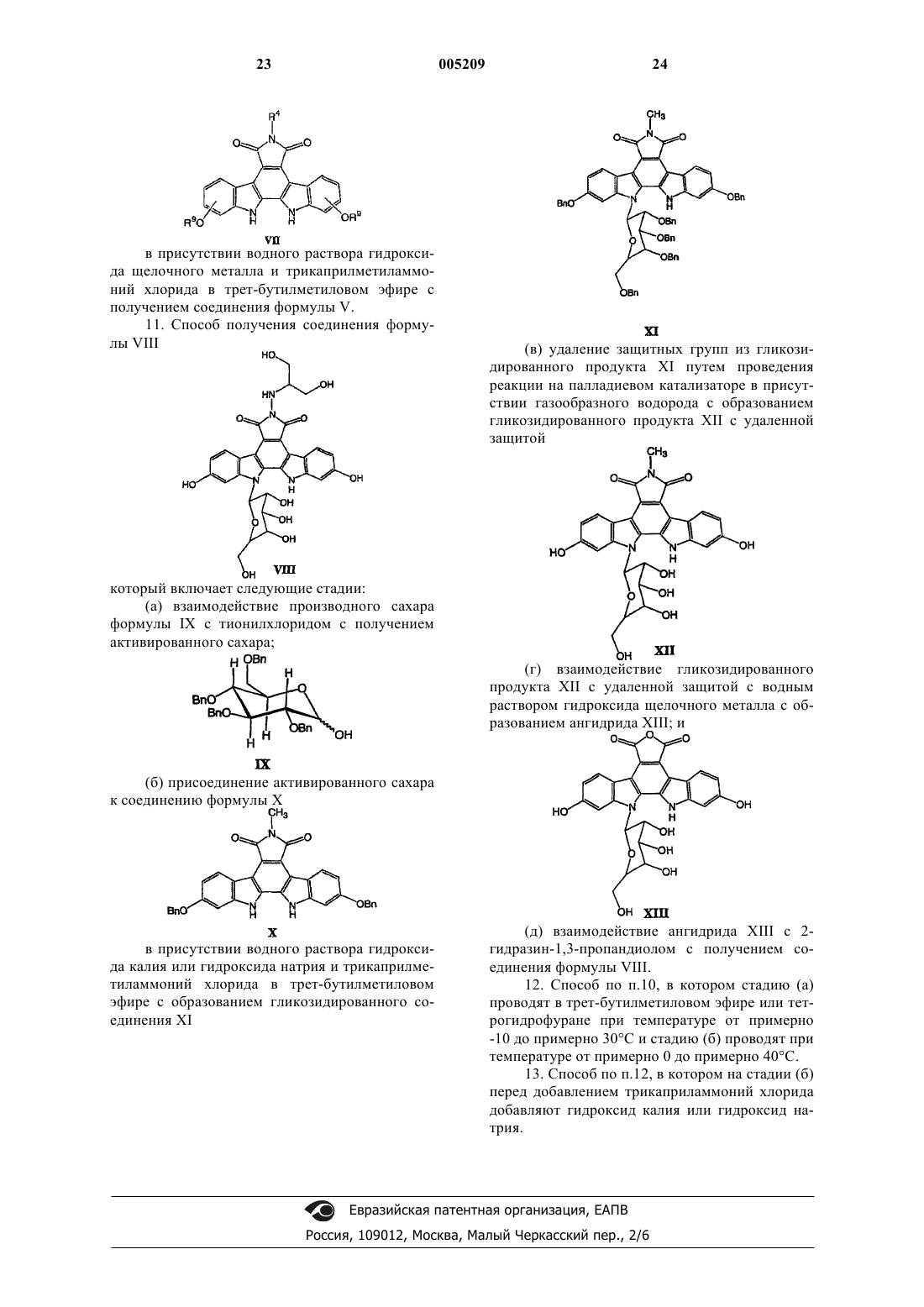
11. Способ получения соединения формулы VIII

который включает следующие стадии:
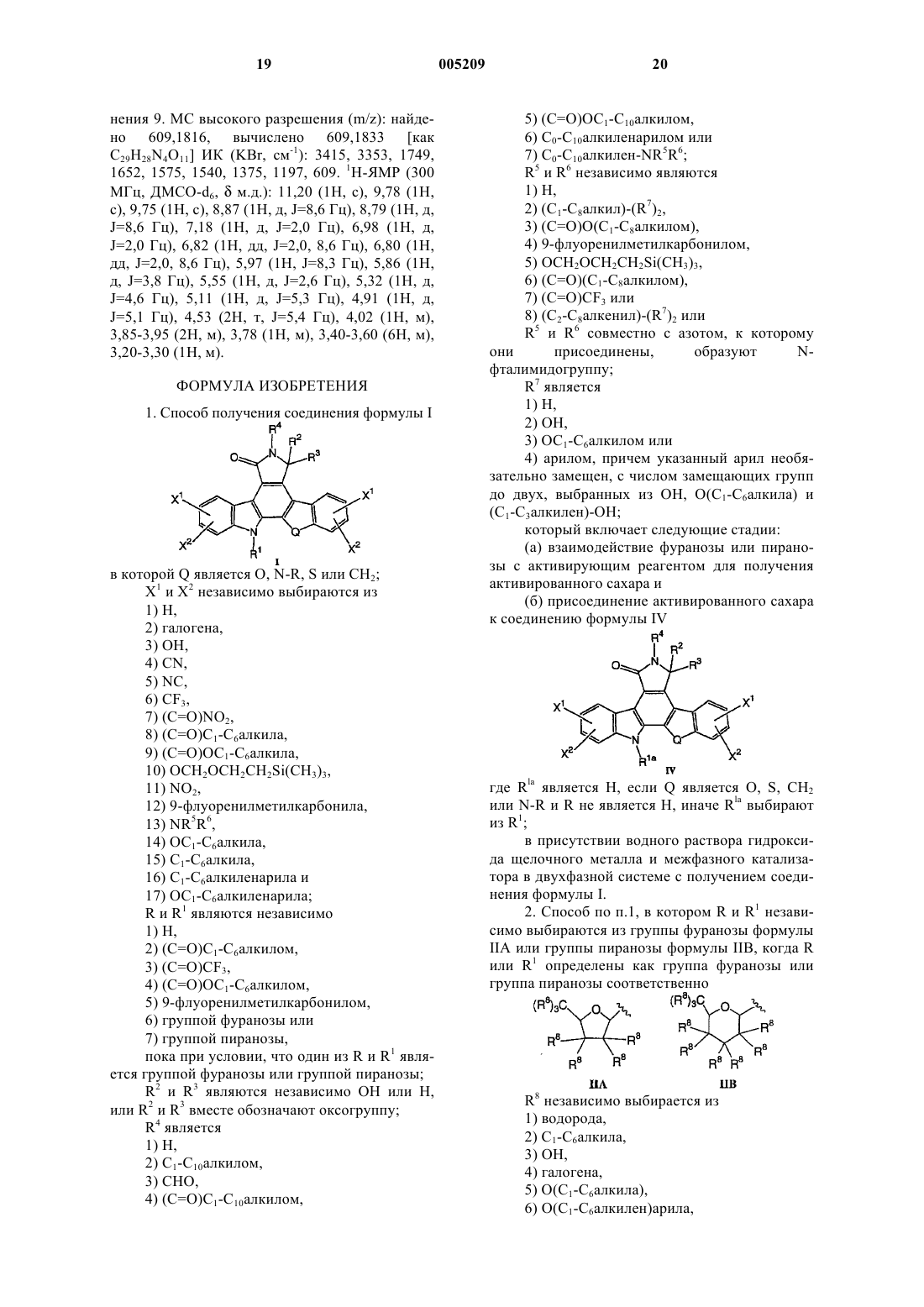
(а) взаимодействие производного сахара формулы IX с тионилхлоридом с получением активированного сахара;

(б) присоединение активированного сахара к соединению формулы X

в присутствии водного раствора гидроксида калия или гидроксида натрия и трикаприлметиламмонии хлорида в трет-бутилметиловом эфире с образованием гликозидированного соединения XI

(в) удаление защитных групп из гликозидированного продукта XI путем проведения реакции на палладиевом катализаторе в присутствии газообразного водорода с образованием гликозидированного продукта XII с удаленной защитой

(г) взаимодействие гликозидированного продукта XII с удаленной защитой с водным раствором гидроксида щелочного металла с образованием ангидрида XIII; и

(д) взаимодействие ангидрида XIII с 2-гидразин-1,3-пропандиолом с получением соединения формулы VIII.
12. Способ по п.10, в котором стадию (а) проводят в трет-бутилметиловом эфире или тетрогидрофуране при температуре от примерно -10 до примерно 30шC и стадию (б) проводят при температуре от примерно 0 до примерно 40шC.
13. Способ по п.12, в котором на стадии (б) перед добавлением трикаприламмоний хлорида добавляют гидроксид калия или гидроксид натрия.
Текст
1 Настоящее изобретение относится к новому способу гликозидирования для получения промежуточных соединений, пригодных для изготовления производных индолпирролкарбазола, ингибирующих рост опухолевых клеток и следовательно пригодных для лечения злокачественных опухолей млекопитающих и им подобных. В области химиотерапии рака уже применяется большое количество соединений в качестве противоопухолевых агентов. Однако существует потребность в дальнейшей разработке более эффективных соединений, действующих против различных новообразований (смотри Труды 47-го Совещания Японского Онкологического Общества, стр.12-15 (1988. Эта потребность привела к разработке производных индолкарбазола (см. патенты США 4487925; 4552842; 4785085; 5591842 и 5922860; патент Японии 20277/91; Journal of Antibiotics,Vol.44, стр. 723-728 (1991); международные патентные заявки WO 91/18003; WO 98/07433; и Европейскую патентную заявку ЕР 0545195 А 1). Показано, что такие соединения действуют как ингибиторы топоизомеразы и следовательно пригодны для лечения злокачественных опухолей (Cancer Chemother. Pharmacol. 34 (suppl):S41-S45 (1994. Успех в лечении большого количества онкологических заболеваний такими соединениями неизбежно влечет за собой разработку усовершенствованных методов для их синтеза (см.Bioorg.Med. Chem. Letters 2000, 10, 419; Tetrahedron 1997, 53, 5937; Tetrahedron 1997, 53,585; и Synthesis 1976, 414). Однако ранее известные методы имеют различные недостатки,такие, например, как применение неудобных растворителей, солей ртути или серебра, низкую эффективность и образование нежелательных побочных продуктов, требующих кропотливых или продолжительных этапов очистки. Таким образом, задачей настоящего изобретения является создание нового подхода к синтезу промежуточных соединений, пригодных для получения противоопухолевых препаратов производных индолпирролкарбазола, в котором преодолены проблемы, свойственные ранее известным способам синтеза. Настоящее изобретение является новым способом гликозидирования для получения промежуточных соединений, пригодных для получения производных индолпирролкарбазола,подобных соединению формулы I, приведенной ниже, ингибирующих рост опухолевых клеток и следовательно пригодных для лечения злокачественных опухолей млекопитающих и им подобных. Реализация настоящего изобретения показана на примере способа приготовления соединения формулы IX1 и X2 независимо выбираются из 1) Н,2) галогена,3) ОН,4) CN,5) NC,6) CF3,7) (C=O)NO2,8) (C=O)C1-C6 алкила,9) (C=O)OC1-C6 алкила,10) OCH2OCH2CH2Si(CH3)3,11) NO2,12) 9-флуоренилметилкарбонила,13) NR5R6,14) OC1-C6 алкила,15) C1-C6 алкила,16) C1-C6 алкиленарила и 17) OC1-C6 алкиленарила;R и R1 являются независимо 1) Н,2) (C=O)C1-C6 алкилом,3) (C=O)CF3,4) (C=O)OC1-C6 алкилом,5) 9-флуоренилметилкарбонилом,6) группой фуранозы или 7) группой пиранозы,пока при условии, что один из R и R1 является группой фуранозы или группой пиранозы;R2 и R3 являются независимо ОН или Н,или R2 и R3 вместе обозначают оксо-группу;R7 является 1) Н,2) ОН,3) OC1-С 6 алкилом или 4) арилом, причем указанный арил необязательно замещен, с числом замещающих групп до двух, выбранных из ОН, O(C1-C6 алкила) и(C1-С 3 алкилен)-ОН; который включает следующие стадии:(а) взаимодействие фуранозы или пиранозы с активирующим реагентом для получения активированного сахара и(б) присоединение активированного сахара к соединению формулы IV где Rla является Н, если Q является О, S, СН 2,или N-R и R не является Н, иначе Rla выбирают из R1; в присутствии водного раствора гидроксида щелочного металла и межфазного катализатора в двухфазной системе с получением соединения формулы I. Другой реализацией является способ, описанный выше, в котором R и R1 независимо выбираются из группы фуранозы формулы IIА или группы пиранозы формулы IIB, когда R или R1 определены как группа фуранозы или группа пиранозы соответственноR8 независимо выбирается из 1) водорода,2) C1-C6 алкила,3) ОН,4) галогена,5) O(C1-C6 алкила) ,6) O(C1-C6 алкилен)арила,7) OSO2(C1-C6 алкила),8) ОSО 2 арила, 005209 4 9) ОСН 2 ОСН 2 СН 2Si(СН 3)3,10) О(С=О) (C1-C6 алкила),11) O(C=O)CF3,12) азидо или 13) NR5R6, или два R8 на одном атоме углерода вместе образуют для обозначения оксогруппу, =N-R5 или=N-R7; и фураноза или пираноза на стадии (а) являются фуранозой формулы IIIA или пиранозой формулы IIIB, соответственно; В другой реализации активирующий реагент на стадии (а) выбирают из галогенангидрида, сульфоната, фосфата, сульфата, бората или ацетата, и двухфазная система на стадии (b) состоит из органических растворителей, выбранных из углеводорода, нитрила, простого эфира,галогенированного углеводорода, кетона, или неполярного апротонного растворителя. Еще одной реализацией является способ,описанный выше, в котором активирующий реагент выбран из SOCl2 или оксалилхлорида. Еще одной реализацией является способ,описанный выше, в котором двухфазная система составлена из метил-трет-бутилового эфира,дихлорметана или трифтортолуола. В еще одной реализации межфазным катализатором на этапе (б) является (Ra)4M+A-;Ra независимо является Н или C1-C18 алифатическим углеводородом; М является N или Р и А является ОН, F, Br, Cl, I, HSO4, CN,MeSO3 или PhCH2CO2. Предпочтительной реализацией является способ, описанный выше, в котором межфазным катализатором является трикаприлметиламмоний хлорид. Другой предпочтительной реализацией является способ, согласно приведенному выше описанию, в котором водный раствор гидроксида щелочного металла на стадии (б) имеет концентрацию от примерно 5 до примерно 95% мас./мас. и гидроксид щелочного металла выбран из гидроксида лития, гидроксида натрия,гидроксида калия и гидроксида цезия. Также предпочтительным является способ,в котором водный раствор гидроксида щелочного металла имеет концентрацию от примерно 45 до примерно 50% мас./об. и гидроксид щелочного металла является гидроксидом калия или гидроксидом натрия. Более предпочтительной реализацией является способ для приготовления соединения формулы V(б) присоединение активированного сахара к соединению формулы VIIR7 является 1) Н,2) ОН,3) OC1-С 6 алкилом или 4) арилом, причем указанный арил необязательно замещен, с числом замещающих групп до двух, выбранных из ОН, О(С 1-С 6 алкила) иR9 является 1) Н,2) C1-С 6 алкилом,3) (C1-С 6 алкилен)арилом,4) SО 2 (C1-С 6 алкилом),5) SO2 арилом,6) CH2OCH2CH2Si(CH3)3,7) (С=O) (C1-C6 алкилом) или 8) (C=O)CF3; который включает следующие стадии:(а) взаимодействие производного сахара формулы VI с хлорангидридом с получением активированного сахара и в присутствии водного раствора гидроксида щелочного металла и трикаприлметиламмоний хлорида в трет-бутилметиловом эфире с получением соединения формулы V. Еще одной предпочтительной реализацией является способ получения соединения формулы VIII который включает следующие стадии:(а) взаимодействие производного сахара формулы IX с тионилхлоридом с получением активированного сахара(б) присоединение активированного сахара к соединению формулы X в присутствии водного раствора гидроксида калия или гидроксида натрия и трикаприлметиламмоний хлорида в трет-бутилметиловом эфире с образованием гликозидированного соединения XI(в) удаление защитных групп из гликозидированного продукта XI путем проведения реакции на палладиевом катализаторе в присутствии газообразного водорода с образованием гликозидированного продукта XII с удаленной защитой(г) взаимодействие гликозидированного продукта XII с удаленной защитой с водным раствором гидроксида щелочного металла с образованием ангидрида XIII(д) взаимодействие ангидрида XIII с 2 гидразин-1,3-пропандиолом с получением соединения формулы VIII. Также предпочтительным является описанный выше способ получения соединения формулы V, в котором стадию (а) проводят в трет-бутилметиловом эфире или тетрагидрофуране при температуре от примерно -10 С до примерно 30 С и стадию (б) проводят при температуре от примерно 0 С до примерно 40 С. И последняя реализация является описанным выше способом, в котором гидроксид калия 8 или гидроксид натрия на стадии (б) добавляется перед трикаприламмоний хлоридом. Соединения настоящего изобретения могут иметь ассиметричные центры, хиральные оси и хиральные плоскости (как это описано в:E.L. Eliel and S.H. Wilen, Stereochemistry of Carbon Compounds, John WileySons, New York,1994, стр.1119-1190) и присутствовать в настоящем изобретении как рацематы, рацемические смеси и как отдельные диастереомеры со всеми возможными изомерами и их смесями,включая оптические изомеры. К тому же, описанные здесь соединения могут существовать как таутомеры, и подразумевается, что обе формы таутомеров входят в объем настоящего изобретения, даже если описана только одна таутомерная структура. Когда любая переменная (например, X1,2 8X , R , R9 и т.д.) встречается более чем один раз в любом компоненте, ее определение в каждом случае является независимым от любого другого случая. Также, комбинации заместителей и переменных являются допустимыми только в тех случаях, если такие комбинации представляют устойчивые соединения. Линии, проведенные от заместителей групп внутрь кольцевых систем, показывают, что указанная химическая связь может быть образована с любым из кольцевых атомов углерода, способным к замещению. Если кольцевая система полициклична,подразумевается, что химическая связь образуется с любым из подходящих атомов углерода только ближайшего кольца. Ясно, что специалистом в данной области техники могут быть выбраны заместители и способы замещения в соединениях настоящего изобретения для получения химически стабильных соединений, легко синтезируемые способами, известными в данной области техники, равно как и способами, приведенными ниже, из широко доступных исходных материалов. В данной работе подразумевается, что "алкил" используется для обозначения разветвленных, линейных и циклических насыщенных алифатических углеводородных групп, имеющих указанное число атомов углерода. Например, C1-С 6, в "C1-C6 алкил" обозначает группы,имеющие 1, 2, 3, 4, 5 или 6 атомов углерода,расположенные в линейном, разветвленном или циклическом порядке. Например, "C1-С 6 алкил" более точно включает метил, этил, пропил, бутил, пентил, гексил и т.д., также как и циклоалкилы, такие как циклопропил, метилциклопропил, диметилциклобутил, циклобутил, циклопентил и циклогексил и т.д. Алкильные заместители могут быть незамещенными или замещенными с числом заместителей от одного до трех, выбранных из галогена, C1-С 6 алкила, ОН,OC1-С 6 алкила,O(C=O)C1-С 6 алкила,О(С=O)OC1-С 6 алкила, амино, амидо, СО 2 Н, CN,NO2, N3, C1-С 6 перфтороалкила и OC1-С 6 перфтороалкила. "Алкокси" обозначает алкильную 9 группу с указанным количеством атомов углерода, соединенных через кислородный мостик. Термин "алкенил" обозначает неароматический углеводородный радикал, линейный,разветвленный или циклический, содержащий от 2 до 10 атомов углерода и по крайней мере одну двойную связь углерод-углерод. Предпочтительно наличие одной двойной связи углеродуглерод и возможно наличие до четырех неароматических двойных связей углерод-углерод. Так, "C2-С 6 алкенил" означает алкенил радикал,содержащий от 2 до 6 атомов углерода. Алкенильные группы включают этенил, пропенил,бутенил, 2-метилбутенил и циклогексенил. Линейные, разветвленные или циклические части алкениловых групп могут содержать двойные связи и могут быть замещенными, если указаны замещенные алкенильные группы. В определенных случаях, заместители могут быть определены с числами углеродных атомов, включающими ноль, как, например, (С 0 С 6) алкилен-NR5R6. Если R5 и R6 являются Н, то в этом случае такое определение может включать NH2, также как и -CH2NH2, -CH2CH2NH2,СН(СН 3)СН 2 СН(СН 3)NН 2, -СН 2 СН (NH2) СН 3 и т.д. В таких случаях подразумевается, что заместители на бивалентных радикалах могут быть присоединены в любой точке и не ограничены терминальной позицией. В данной работе подразумевается, что"арил" используется для обозначения замещенных и незамещенных фенила или нафтила. В случае замещенного арила, число заместителей может быть от одного до трех, выбранных из галогена, C1-С 6 алкила, ОН, OC1-С 6 алкила,O(C=O)C1-С 6 алкила, О(С=О)ОC1-С 6 алкила,амино, амидо, СО 2 Н, CN, NO2, N3, C1-С 6 перфторалкила и OC1-С 6 перфторалкила. Как это принято специалистами в данной области техники, применяемые в настоящей работе термины "гало" или "галоген" обозначают хлор, фтор, бром и йод. При использовании таких определений,как "(C1-С 8 алкил)-(R7)2", подразумевается, что переменная R7 присоединяется в любом месте алкильной цепи. Следовательно, если R7 определяется как ОН, то в этом случае определение может включать следующее: СН 2 ОН,СН 2 СН 2 ОН, СН(СН 3)СН(ОН)СН 3, СН(СН 3)СН(ОН) СН 2-СН(ОН)СН 3 и т.д. Термины "алкилен" и "алкенилен" являются простой ссылкой соответственно на алкильную или алкенильную группы, определенные выше, содержащие указанное количество атомов углерода и являющиеся двухвалентными. Например, "C1-C4 алкилен" включает -СН 2-,-СН 2 СН 2-, -СН(СН 3)СН 2- и т.д. Определения R и R1 включают производные сахаров фуранозы и пиранозы. Предпочтительными производными сахаров являются пиранозы с защищенным кислородом, такие как Dглюкопираноза; 6-дезокси-6,6-дифтор-D-глюко 005209 10 пираноза; 6-дезокси-6-азидо-D-глюкопираноза; 6-амино-6-дезокси-D-глюкопираноза; 6-азидоD-глюкопираноза; 6-амино-D-глюколираноза; 4 дезокси-4,4-дифтор-6-дезокси-6-азидо-D-глюкопираноза; 2-фтор-D-глюкопираноза; D-галактопираноза; 4-дезокси-D-галактопираноза; 4-дезокси-D-глюкопираноза; и 4-метокси-D-глюкопираноза (см., например, WO 97/07433, включенное по ссылке). Предпочтительные фуранозы включают ксилофуранозу, арабинофуранозу,рибофуранозу, аллофуранозу и 2-дезоксирибофуранозу. В общем случае R9 может быть любой известной, защищающей кислород группой. Примеры таких защитных групп включают, но не ограничены следующими: бензил, паранитробензил, толил и им подобные. Более предпочтительной защитной группой является бензил (Вn), т.е. СН 2 Рh. Другие подходящие защитные группы должны быть известны специалистам в данной области техники, примеры могут быть найдены в Protective Groups in Organic"двухфазная система" используется для обозначения двухфазной системы растворителя, состоящей из водной фазы и органической фазы. Реагент, активирующий сахар для присоединения, может быть без труда выбран специалистом в данной области техники. Примеры таких реагентов включают галогенангидриды(такие как SOCl2, POCl3, SOBr2, РОВr3, РВr3 и оксалилхлориды), сульфонилгалогенид и т.п. Предпочтительными реагентами являются тионилхлорид и оксалилхлорид. Наиболее предпочтительным является тионилхлорид. Другие применяемые реагенты активации включают трифенилфосфин/I2 и трифенилфосфин/азидодикарбоксилат. Подходящий растворитель для применения в реакции активирования сахара может быть определен любым квалифицированным химиком. Предпочтительными растворителями являются углеводороды (например, толуол, ксилол, гептан и гексан), нитрилы (например, ацетонитрил),эфиры(например,третбутилметиловый эфир и тетрагидрофуран), галогенированные углеводороды (например, метиленхлорид,тетрахлорметан,хлороформ,трифтортолуол и дихлорбензол), кетоны (например, метилизобутилкетон и ацетон) и неполярные апротонные растворители (например,N,N-диметилформамид и 1-метил-2-пирролидинон). Наиболее предпочтительным растворителем является трет-бутилметиловый эфир. Реакция активации может проходить при температурах в пределах от примерно -50 С до примерно 200 С. Предпочтительной является температура от примерно -10 С до примерно 30 С. 11 Также, специалист в данной области техники сможет легко подобрать подходящий растворитель для использования в двухфазной реакции присоединения. Подходящие растворители включают углеводороды (такие как толуол,ксилол, гептан и гексан), нитрилы (такой как ацетонитрил), эфиры (такие как третбутилметиловый эфир и тетрагидрофуран), галогенированные углеводороды (такие как метиленхлорид, тетрахлорметан, хлороформ, трифтортолуол и дихлорбензол), кетоны (такие как метилизобутилкетон и ацетон) и неполярные апротонные растворители (такие как N,Nдиметилформамид и 1-метил-2-пирролидинон). Наиболее предпочтительными растворителями являются трет-бутилметиловый эфир, дихлорметан, трифтортолуол. Реакция присоединения может проходить при температуре в пределах от примерно -50 С до примерно 200 С. Наиболее предпочтительной является температура от примерно 0 С до примерно 40 С. Предпочтительными основаниями для реакции присоединения являются гидроксиды щелочных металлов, такие как гидроксиды лития, натрия, калия и цезия. Наиболее предпочтительными являются гидроксид калия и гидроксид натрия. Концентрация основания в воде может изменяться от примерно 5% мас./мас. до примерно 95% мас./мас. Более предпочтительными концентрациями являются от примерно 45 до примерно 50% мас./мас. Предпочтительными межфазными реагентами в реакции присоединения являются реагенты с общей формулой (Ra) 4M+A-, где Ra независимо Н или C1-C18 алифатический углеводород; М является N или Р; и А является ОН, F,Br, Cl, I, HSO4, CN, MeSO3 или PhCH2CO2. Предпочтительным межфазным катализатором является трикаприлметиламмоний хлорид. Другие подходящие межфазные катализаторы включают, но не ограничивается трис-[2-(2 метоксиэтокси) этил]амин (TDA-1) ; BnEt3N+Cl-; и (Bu) 3NH+HSO4-. Краткое описание схем Схема А иллюстрирует один из возможных обобщенных подходов к получению гликозидированного субстрата А-6. В данной области техники известны другие подходы, некоторые из них описаны Kojiri и другими в патентной заявке США 5922860 (поданной 13 июля 2000 г.) и включенной здесь по ссылке. Схема В показывает гликозидирование А-6 с использованием межфазного катализа с целью получения промежуточных соединений типа В-3. Схемы С и D показывают возможные дальнейшие модификации для получения соединений, известных в качестве ингибиторов топоизомеразы. Примеры Представленные примеры предназначены для того, чтобы дополнительно облегчить понимание изобретения. Конкретные используемые материалы, типы и состояния предназначены для дополнительной иллюстрации изобретения, а не с целью ограничения его объема. Промежуточное вещество 5, используемое в реакции гликозидирования согласно настоящему изобретению, может быть получено способом, описанным Kojiri и другими в патентной заявке США 5922860 (поданной 13 июля 2000 г.), включенной здесь по ссылке. В общих чертах эта процедура описана ниже в примерах с 1 по 5. Пример 1. Получение соединения, представленного формулой 1 284 г бензилоксииндола было растворено в 3 л ТГФ (тетрагидрофуран) и к этому добавлено 2,7 л гексаметилдисилазида лития (как 1 М раствор в ТГФ). Затем смесь перемешивалась в атмосфере азота при -10 С в течение 45 мин и к 14 ней по каплям в течение часа добавлялось 3 л раствора ТГФ, содержащего 340 г 2,3-дибромN-метилимида малеиновой кислоты. После окончания добавления полученная смесь перемешивалась в течение 15 мин при 0 С. Реакционная смесь выливалась в 10 л 2 н. соляной кислоты и экстрагировалась 30 л этилацетата. Органический слой был промыт насыщенным водным раствором гидрокарбоната натрия и затем насыщенным водным раствором хлорида натрия, высушен и упарен. Полученный осадок был перекристаллизован из метанола с получением требуемого соединения 1. МС высокого разрешения (m/z): найдено 410,0292, вычислено 410,0266 [как C20H15N2O3Br] ИК (KВr, см-1): 3330, 3318, 1762, 1701, 1606, 1511, 1450, 1165,1135, 1041, 794. 1 Н-ЯМР (300 МГц, CDCl3,м.д.): 8,60 (1 Н, шир.с), 7,96 (1 Н, д, J=8,1 Гц),7,94 (1 Н, д, J=2,5 Гц), 7,33-7,47 (5 Н, м), 7,00 (1 Н,дд, J=2,5, 8,8 Гц), 6,97 (1 Н, д, J=2,5 Гц), 5,13 1,00 г соединения 1, полученного в примере 1, 637 мг ди-трет-бутилдикарбоната и 3 мг 4N,N-диметиламинопиридина было растворено в 200 мл ТГФ, и этот раствор перемешивался при комнатной температуре в течение часа. Затем реакционная смесь была упарена, полученный остаток был перекристаллизован из этилацетатгексана с получением требуемого соединения(2). ИК (KВr, см-1): 1740, 1714, 1614, 1527, 1487,1443, 1373, 1227, 1153. МС высокого разрешения (m/z): найдено 510,0771, вычислено 510,0791 [как C25H23N2O5Br] 1H-ЯМР (300 МГц,CDCl3,м.д.): 8,10 (1 Н, с), 7,91 (1 Н, д, J=2,3 Гц), 7,73 (1 Н, д, J=8,9 Гц), 7,34-7,50 (5 Н, м) ,7,03 (1 Н, дд, J=2,3, 8,5 Гц), 5,16 (2 Н, с), 3,18 (3 Н,с), 1,68 (9 Н, с). Пример 3. Получение соединения, представленного формулой 3 218,4 мг 6-бензилоксииндола было растворено в 20 мл ТГФ и к этому добавлено 2,35 мл гексаметилдисилазида лития (как 1 М раствор 15 ТГФ). Затем смесь перемешивалась в атмосфере азота при 0 С в течение 15 мин, к ней по каплям в течение 10 мин добавлялось 10 мл раствора ТГФ, содержащего 500 мг соединения (2), полученного в примере 2. После завершения добавления полученная смесь перемешивалась при комнатной температуре в течение 0,5 ч. Реакционная смесь выливалась в 100 мл 2 н. соляной кислоты и экстрагировалась 400 мл этилацетата. Органический слой был промыт водой, насыщенным водным раствором гидрокарбоната натрия и затем насыщенным водным раствором хлорида натрия, высушен и упарен. Полученный осадок был перекристаллизован из толуолгексана с получением требуемого соединения(3). МС высокого разрешения (m/z): найдено 653,2556, вычислено 653,2526 [как C40H35N3O6] ИК (КВr, см-1): 1740, 1701, 1646, 1623, 1543,1445, 1155. 1H-ЯМР (300 МГц, CDCl3,м.д.): 8,41 (1 Н, шир.с), 7,97 (1 Н, с), 7,84 (1 Н, шир.с),7,68 (1 Н, шир.с), 7,16-7,43 (10 Н, м) , 6,98 (1 Н, д,J=9,2 Гц), 6,85 (1 Н, шир.с), 6,74 (1 Н, д, J=9,2 Гц), 6,58 (1 Н, д, J=9,2 Гц), 6,52 (1 Н, д, J=9,2 Гц),5,05 (2 Н, с), 5,02 (2 Н, с), 3,19 (3 Н, с), 1,67 (9 Н,с). Пример 4. Получение соединения, представленного формулой 4(как 40% раствор в метаноле), и этот раствор перемешивался при комнатной температуре в течение 30 мин. Затем реакционная смесь была упарена, полученный осадок был перекристаллизован из смеси дихлорметан-ацетон-гексан с получением 68,6 мг требуемого соединения (4). МС высокого разрешения (m/z): найдено 553,1982, вычислено 553,2002 [как C35H27N3O4] ИК (KВr, см-1): 3419, 3350, 1759, 1697, 1620,1533, 1454, 1383, 1292, 1167. 1H-ЯМР (300 МГц,ДMCO-d6,м.д.): 11,48 (2 Н, с), 7,62 (2 Н, с),7,28-7,45 (10 Н, м), 6,95 (2 Н, д, J=l,2 Гц), 6,70 16 бензохинона были растворены в 50 мл толуола,и этот раствор перемешивался при 110 С в течение 40 мин. После остывания реакционной смеси до комнатной температуры нерастворимые вещества были отфильтрованы и промыты 30 мл метанола. Остаток был перекристаллизован из диметилсульфоксид-дихлорметанметанола с получением требуемого соединения(5). МС высокого разрешения (m/z): найдено 551,1829, вычислено 551,1845 [как C35H25N3O4] ИК (KВr, см-1): 3257, 1740, 1675, 1620, 1571,1402, 1246, 1178. 1 Н-ЯМР (300 МГц, ДМСО-d6, м.д.): 11,46 (2 Н, с), 8,79 (2 Н, д, J=8,5 Гц), 7,53N,N-диметиформамида при 23 С и затем охлаждено до 9 С. Тионилхлорид (16,2 мл; 222 моль) добавлялся медленно в течение 15 мин, за это время температура поднялась до 20 С. Раствор был нагрет до примерно 30 С и выдержан в течение часа. Затем раствор был охлажден до-10 С и к нему был добавлен 10% мас./мас. КОН(около 150 мл), в течение этого времени температура не превышала 0 С. Раствор был нагрет до 22 С. Водный слой был экстрагирован третбутилметиловым эфиром (МТБЭ) (1x300 мл). Затем все органические слои были промыты соляным раствором (1x150 мл) и водой (1x200 мл). Раствор был упарен при пониженном давлении до 350 мл и использовался на следующем этапе без дополнительной очистки. Этап 2 72 г (131 ммоль) соединения 5 из примера 5 было растворено в 600 мл МТБЭ и перемешивалось в течение 10 мин при 23 С. Затем был добавлен раствор 6-2, приготовленный на стадии 1, и через 10 мин был добавлен 45% мас./мас. водный раствор КОН (300 мл). По истечении 10 мин медленно в течение 22 мин был добавлен 40% мас./мас. Aliquat 336 (72 г в 110 г МТБЭ). Aliquat 336 является торговой маркой трикаприлметиламмоний хлорида, поставляемого Aldrich Chemical Co., Inc., in Milwaukee, Wis 17consin. Раствор был выдержан при 23 С в течение 6 ч, затем было добавлено 350 мл воды и в течение 5 мин проводилось пассивное перемешивание. Слои были разделены и водный слой промыт МТБЭ (1x300 мл). Все органические слои были затем промыты 10% мас./мас. лимонной кислотой (1x300 мл) и водой (1x300 мл). Органический слой перемешивался при 22 С в течение ночи, в этот период началась кристаллизация продукта (6-3). Затем раствор был упарен при атмосферном давлении (точка кипения 55 С) до 625 мл. На этой фазе раствор был охлажден до 23 С, и медленно в течение 1 ч добавлялся метанол (225 мл). Затем суспензия была охлаждена до -5 С и выдержана в течение 45 мин. Сухой остаток был отделен и промыт охлажденным 1:1 метанол/МТБЭ (2x400 мл). После сушки в вакууме при 25-40 С был получен продукт 6-3 с чистотой более 99%, определенной методом жидкостной хроматографии. Следующий пример, заимствованный из патентной заявки США Kojiri и другие 5922860 и ранее включенной по ссылке, иллюстрирует применение продуктов гликозидирования в синтезе известного ингибитора топоизомеразы (9). Пример 7. Получение соединения, представленного формулой 7 100 мг соединения 6-3 было растворено в 6 мл хлороформ-метанола (2:1) и к этому раствору была добавлена палладиевая чернь в количестве, необходимом для катализа. Эта смесь перемешивалась в течение 2 ч в атмосфере водорода. Затем катализатор был отфильтрован,фильтрат был упарен. Полученный осадок был кристаллизован из метанол-ацетон-этилацетатгексана, производства Sephadex LH-20, элюирован при помощи хлороформ-метанол-этанолтетрагидрофурана (5:2:2:1) и перекристаллизован из ацетон-метанол-гексана для получения требуемого соединения (7). МС высокого разрешения (m/z): найдено 533,1429, вычислено 533,1434 [как C27H23N3O9] ИК (KВr, см-1): 3328,1733, 1683, 1678, 1540, 1417, 1126, 1081, 611. 1HЯМР (300 МГц, ДМСО-d6,м.д.): 11,20 (1 Н, с),9,76 (1 Н, с), 9,74 (1 Н, с), 8,88 (1 Н, д, J=8,6 Гц),8,80 (1 Н, д, J=8,6 Гц), 7,18 (1 Н, д, J=2,l Гц), 6,99 1,2 г соединения (7) было растворено в 40 мл 10% водного раствора гидроксида калия и этот раствор перемешивался в течение часа при комнатной температуре. Реакционная смесь была нейтрализована путем добавления 40 мл 2 н. соляной кислоты и затем экстрагирована 1 л метилэтилкетона. Органический слой был промыт насыщенным водным раствором хлорида натрия, высушен и упарен. Полученный осадок был перекристаллизован из ацетон-гептана для получения требуемого соединения (8). МС высокого разрешения (m/z): найдено 520,1147, вычислено 520,1118 [как C26H20N2O10] ИК (KВr,см-1): 3311, 1810, 1739, 1652, 1626, 1558, 1405,1091, 611. 1H-ЯМР (300 МГц, ДMCO-d6,м.д.): 11,4 (1 Н, с), 9,95 (1 Н, с), 9,92 (1 Н, с), 8,69 (1 Н, д,J=7,7 Гц), 8,63 (1 Н, д, J=7,7 Гц), 7,25 (1 Н, д,J=1,5 Гц), 7,03 (1 Н, д, J=1,5 Гц), 6,90 (1 Н, дд,J=1,5, 7,7 Гц), 6,87 (1 Н, д, J=1,5, 7,7 Гц), 6,06 500 мг соединения 8 было растворено в 50 мл N,N-диметилформамида, к этому было добавлено 152 мг 2-гидразин-1,3-пропандиола. Эта смесь перемешивалась при 80 С в течение 1 ч. Затем реакционная смесь была упарена, полученный осадок был очищен при помощиSephadex LH-20 (хлороформ-метанол-этанолвода=5:2:2:1) с получением требуемого соеди 19 нения 9. МС высокого разрешения (m/z): найдено 609,1816, вычислено 609,1833 [какC29H28N4O11] ИК (KВr, см-1): 3415, 3353, 1749,1652, 1575, 1540, 1375, 1197, 609. 1 Н-ЯМР (300 МГц, ДМСО-d6,м.д.): 11,20 (1 Н, с), 9,78 (1 Н,с), 9,75 (1 Н, с), 8,87 (1 Н, д, J=8,6 Гц), 8,79 (1 Н, д,J=8,6 Гц), 7,18 (1 Н, д, J=2,0 Гц), 6,98 (1 Н, д,J=2,0 Гц), 6,82 (1 Н, дд, J=2,0, 8,6 Гц), 6,80 (1 Н,дд, J=2,0, 8,6 Гц), 5,97 (1 Н, J=8,3 Гц), 5,86 (1 Н,д, J=3,8 Гц), 5,55 (1 Н, д, J=2,6 Гц), 5,32 (1 Н, д,J=4,6 Гц), 5,11 (1 Н, д, J=5,3 Гц), 4,91 (1 Н, д,J=5,1 Гц), 4,53 (2 Н, т, J=5,4 Гц), 4,02 (1 Н, м),3,85-3,95 (2 Н, м), 3,78 (1 Н, м), 3,40-3,60 (6 Н, м),3,20-3,30 (1 Н, м). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы IX1 и X2 независимо выбираются из 1) Н,2) галогена,3) ОН,4) CN,5) NC,6) CF3,7) (C=O)NO2,8) (С=О)С 1-С 6 алкила,9) (C=O)OC1-C6 алкила,10) OCH2OCH2CH2Si(СH3)3,11) NO2,12) 9-флуоренилметилкарбонила,13) NR5R6,14) OC1-С 6 алкила,15) C1-С 6 алкила,16) C1-С 6 алкиленарила и 17) OC1-С 6 алкиленарила;R и R1 являются независимо 1) Н,2) (C=O)C1-C6 алкилом,3) (C=O)CF3,4) (C=O)OC1-C6 алкилом,5) 9-флуоренилметилкарбонилом,6) группой фуранозы или 7) группой пиранозы,пока при условии, что один из R и R1 является группой фуранозы или группой пиранозы;R2 и R3 являются независимо ОН или Н,или R2 и R3 вместе обозначают оксогруппу;R7 является 1) Н,2) ОН,3) OC1-С 6 алкилом или 4) арилом, причем указанный арил необязательно замещен, с числом замещающих групп до двух, выбранных из ОН, О(C1-С 6 алкила) и(C1-С 3 алкилен)-ОН; который включает следующие стадии:(а) взаимодействие фуранозы или пиранозы с активирующим реагентом для получения активированного сахара и(б) присоединение активированного сахара к соединению формулы IV где Rla является Н, если Q является О, S, СН 2 или N-R и R не является Н, иначе Rla выбирают из R1; в присутствии водного раствора гидроксида щелочного металла и межфазного катализатора в двухфазной системе с получением соединения формулы I. 2. Способ по п.1, в котором R и R1 независимо выбираются из группы фуранозы формулыIIА или группы пиранозы формулы IIB, когда R или R1 определены как группа фуранозы или группа пиранозы соответственноR8 независимо выбирается из 1) водорода,2) C1-С 6 алкила,3) ОН,4) галогена,5) O(C1-C6 алкила),6) O(C1-C6 алкилен)арила, 21 7) OSO2(C1-С 6 алкила),8) OSO2 арила,9) OCH2OCH2CH2Si(CH3)3,10) О(С=О)(С 1-С 6 алкила),11) O(C=O)CF3,12) азидо или 13) NR5R6 или два R8 на одном атоме углерода вместе образуют оксогруппу, =N-R5 или =N-R7; и фураноза или пираноза на стадии (а) являются фуранозой формулы IIIА или пиранозой формулы IIIB соответственно 3. Способ по п.2, в котором активирующий реагент на стадии (а) выбирают из галогенангидрида, и двухфазная система на стадии (b) включает органический растворитель, выбранный из углеводорода, нитрила, простого эфира,галогенированного углеводорода, кетона или неполярного апротонного растворителя. 4. Способ по п.3, в котором активирующий реагент выбирают из SOCl2 или оксалилхлорида. 5. Способ по п.3, в котором двухфазная система составляется из метил-трет-бутилового эфира, дихлорметана или трифтортолуола. 6. Способ по п.3, в котором межфазным катализатором на стадии (б) является (Ra)4M+A-;Ra независимо является Н или C1-С 18 алифатическим углеводородом; М является N или Р; и А является ОН, F, Br, Cl, I, HSO4, CN,MeSO3 или PhCH2CO2. 7. Способ по п.6, в котором межфазным катализатором является трикаприлметиламмоний хлорид. 8. Способ по п.3, в котором водный раствор гидроксида щелочного металла на стадии(б) имеет концентрацию от примерно 5 до примерно 95% мас./мас. и гидроксид щелочного металла выбран из гидроксида лития, гидроксида натрия, гидроксида калия и гидроксида цезия. 9. Способ по п.8, в котором водный раствор гидроксида щелочного металла имеет концентрацию от примерно 45 до примерно 50% мас./об. и гидроксид щелочного металла является гидроксидом калия или гидроксидом натрия. 10. Способ получения соединения формулы VR7 является 1) Н,2) ОН,3) OC1-С 6 алкилом или 4) арилом, причем указанный арил необязательно замещен, с числом замещающих групп до двух выбранных из ОН, О(С 1-С 6 алкила) иR9 является 1) Н,2) C1-С 6 алкилом,3) (C1-С 6 алкилен)арилом,4) SО 2(С 1-С 6 алкилом),5) SO2 арил,6) CH2OCH2CH2Si(CH3)3,7) (С=O)(C1-С 6 алкилом) или 8) (C=O)CF3; который включает следующие стадии:(а) взаимодействие производного сахара формулы VI с хлорангидридом с получением активированного сахара и(б) присоединение активированного сахара к соединению формулы VII в присутствии водного раствора гидроксида щелочного металла и трикаприлметиламмоний хлорида в трет-бутилметиловом эфире с получением соединения формулы V. 11. Способ получения соединения формулы VIII(в) удаление защитных групп из гликозидированного продукта XI путем проведения реакции на палладиевом катализаторе в присутствии газообразного водорода с образованием гликозидированного продукта XII с удаленной защитой который включает следующие стадии:(а) взаимодействие производного сахара формулы IX с тионилхлоридом с получением активированного сахара;(г) взаимодействие гликозидированного продукта XII с удаленной защитой с водным раствором гидроксида щелочного металла с образованием ангидрида XIII; и(б) присоединение активированного сахара к соединению формулы X в присутствии водного раствора гидроксида калия или гидроксида натрия и трикаприлметиламмоний хлорида в трет-бутилметиловом эфире с образованием гликозидированного соединения XI(д) взаимодействие ангидрида XIII с 2 гидразин-1,3-пропандиолом с получением соединения формулы VIII. 12. Способ по п.10, в котором стадию (а) проводят в трет-бутилметиловом эфире или тетрогидрофуране при температуре от примерно-10 до примерно 30 С и стадию (б) проводят при температуре от примерно 0 до примерно 40 С. 13. Способ по п.12, в котором на стадии (б) перед добавлением трикаприламмоний хлорида добавляют гидроксид калия или гидроксид натрия.
МПК / Метки
МПК: A61K 31/33
Метки: межфазного, гликозидирование, катализа, применением, индолкарбазола
Код ссылки
<a href="https://eas.patents.su/13-5209-glikozidirovanie-indolkarbazola-s-primeneniem-mezhfaznogo-kataliza.html" rel="bookmark" title="База патентов Евразийского Союза">Гликозидирование индолкарбазола с применением межфазного катализа</a>
Способ предотвращения коррозии металлов с применением силанов

Номер патента: 1588
Опубликовано: 25.06.2001
Авторы: Жанг Чумбин, Ван Оэй Вим Дж., Субраманиан Виджай
МПК: C23C 22/56, B05D 7/14, C09D 4/00...
Метки: применением, металлов, способ, коррозии, предотвращения, силанов
Формула / Реферат:
1. Способ обработки металлической подложки, включающий стадии: (a) обеспечения металлической подложки; (b) нанесения покрытия из первого обрабатывающего раствора непосредственно на поверхность металла, причем первый обрабатывающий раствор содержит, по меньшей мере, 0,1% по объему, по меньшей мере, одного силана, где силаном является многофункциональный силан, имеющий, по меньшей мере, две тризамещенные силильные группы, в которых заместители...
Способ гидроформилирования с применением многоступенчатых реакторов

Номер патента: 1834
Опубликовано: 27.08.2001
Авторы: Биллиг Эрнст, Николсон Джеймс Клэйр, Брайант Дэвид Роберт, Баннинг Дональд Лерой, Бекер Майкл Карл
МПК: B01J 19/18, C07C 45/50
Метки: многоступенчатых, применением, реакторов, гидроформилирования, способ
Формула / Реферат:
1. Способ гидроформилирования одного или более продуктов в многоступенчатом реакторе, причём способ включает реакцию в указанном многоступенчатом реакторе одного или более реагентов с монооксидом углерода в присутствии металлокомплексного катализатора с фосфорорганическим лигандом и необязательно свободного фосфорорганического лиганда с получением одного или более продуктов, при таких условиях, в которых указанный металлокомплексный катализатор...
Способ металлообработки с применением смазки

Номер патента: 1309
Опубликовано: 26.02.2001
Автор: Баллиетт Роберт В.
МПК: B21B 45/02, B21C 43/00
Метки: металлообработки, применением, смазки, способ
Формула / Реферат:
1. Способ металлообработки, включающий смазывание обрабатываемого металла фторированной, инертной жидкостью, выбранной из группы, состоящей из алифатических перфторкарбоновых жидкостей общей формулы СnF2n+2; пeрфторморфолинов общей формулы CnF2n+1ON, пeрфтораминов высокофторированных аминов, перфторэфиров, высокофторированных эфиров, и продуктов их полимеризации, при этом указанные фторированные инертные жидкости используют в замещенном и...
Способ кристаллизации ингибитора обратной транскриптазы с применением противорастворителя

Номер патента: 1805
Опубликовано: 27.08.2001
Авторы: Кларк Уилльям, Кукура Джозеф Л., Крокер Луис С.
МПК: C07D 265/18
Метки: транскриптазы, обратной, противорастворителя, ингибитора, применением, кристаллизации, способ
Формула / Реферат:
1. Способ кристаллизации соединения структурной формулы включающий следующие стадии: (1) растворение соединения в растворителе при соотношении от около 3,0 мл до около 10,0 мл растворителя на 1 г соединения; (2) фильтрацию раствора соединения для удаления твердых частиц вещества; (3) добавление к перемешиваемому раствору противорастворителя в течение периода времени от около 30 мин до около 1 ч при комнатной температуре для достижения точки...
Способы лечения женщин в постклимактерическом состоянии с применением ультранизких доз эстрогена

Номер патента: 4573
Опубликовано: 24.06.2004
Авторы: Еллмэн Херман, Каммингс Стивен, Еттингер Брюс
МПК: A61K 31/565, A61P 15/12
Метки: эстрогена, женщин, лечения, доз, способы, состоянии, применением, ультранизких, постклимактерическом
Формула / Реферат:
1. Способ лечения соматических физических состояний во время постклимактерического периода, отличающийся тем, что он предусматривает системное введение эстрогена в количестве, достаточном для обеспечения уровня эстрогена в сыворотке, эквивалентного содержанию в сыворотке эстрадиола, равного примерно 5-15 пг/мл. 2. Способ по п.1, отличающийся тем, что физические состояния выбраны из группы, включающей постклимактерическое уменьшение содержания...
Предыдущий патент: Фунгицидная смесь на основе амидных соединений и производных арилоксихинолина
Следующий патент: Сульфонамиды гидроксидифенилмочевины в качестве антагонистов рецептора ил-8
Случайный патент: Контроллер шагового двигателя и игровой автомат