Вакцина для специфической профилактики гриппа
Номер патента: 16866
Опубликовано: 30.08.2012
Авторы: Катлинский Владимир Антонович, Катлинский Антон Викентьевич, Семченко Андрей Викторович, Мельников Сергей Яковлевич, Коровкин Сергей Анатольевич











Формула / Реферат
Вакцина для профилактики гриппа, представляющая собой смесь поверхностных и внутренних антигенов вирусов гриппа типа А и В, полученная расщеплением очищенных вирионов детергентом октилгликозидом, в которой поверхностные антигены представлены в виде виросом размером 100-150 нм, включающих в свой состав собственные вирусные фосфолипиды, и внутренние антигены представлены в виде мицелл, причем соотношение гемагглютинина к внутренним антигенам составляет 1:(0,5-2,5), а к вирусным фосфолипидам составляет 1:(2-4).
Текст
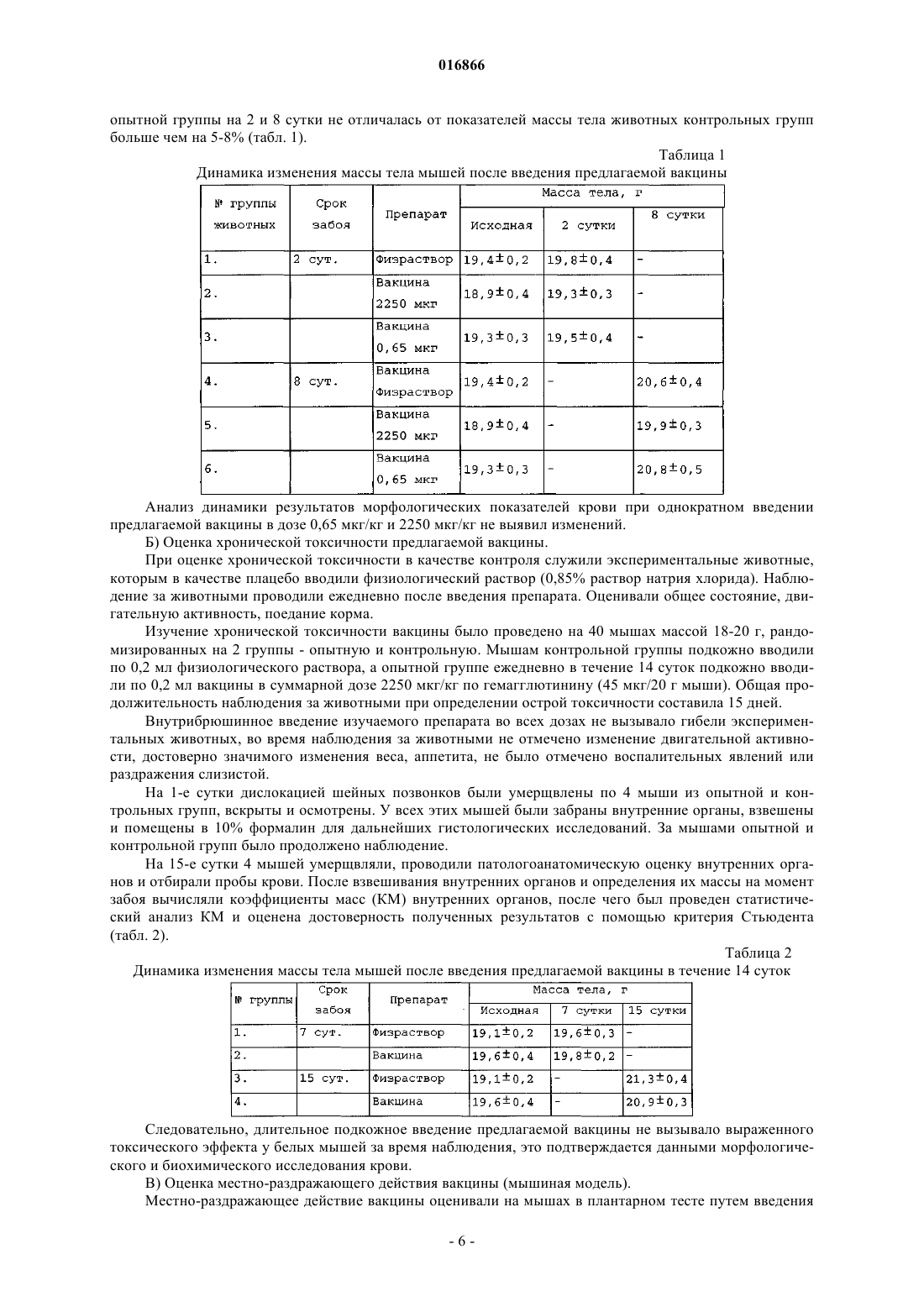
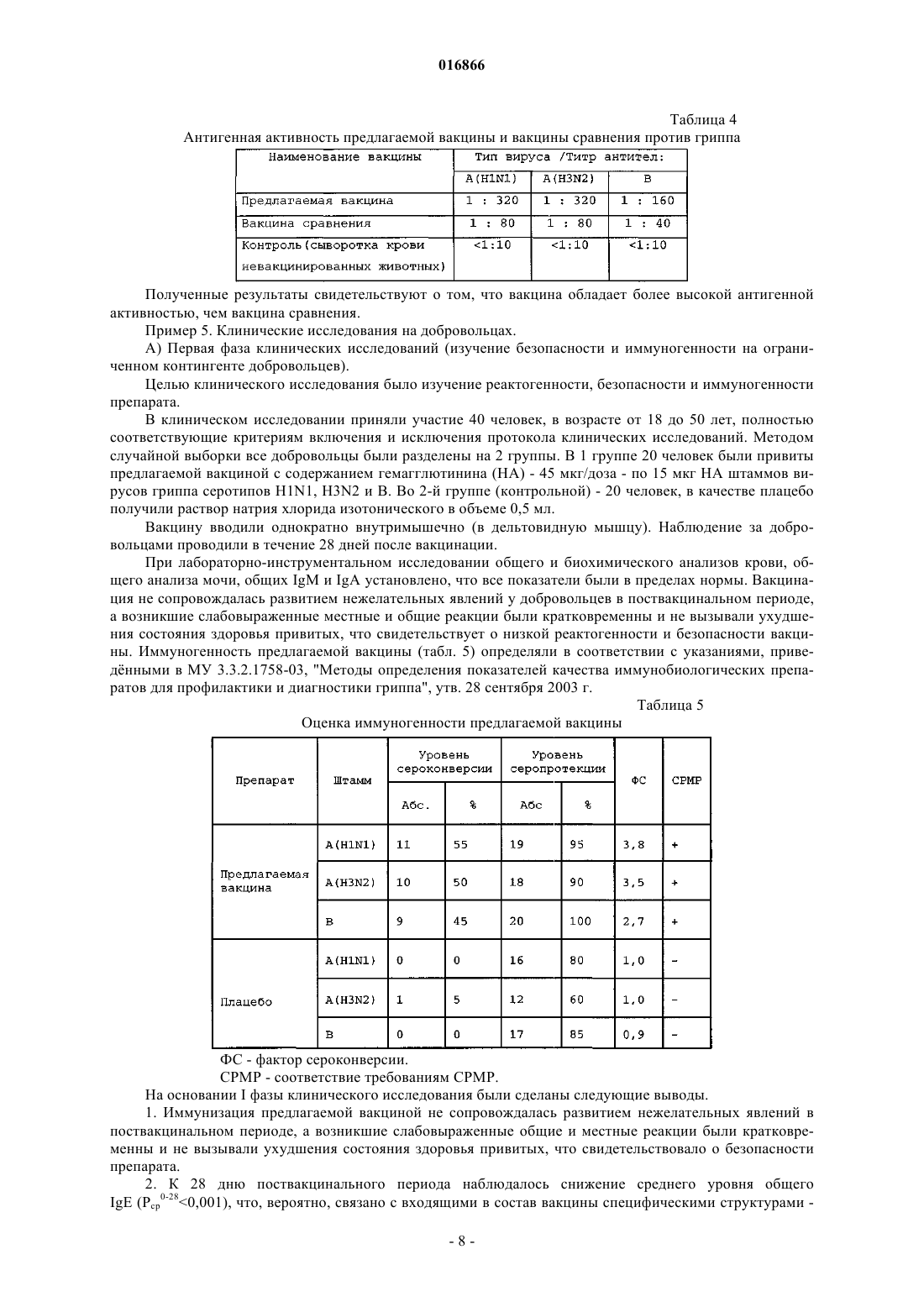
ВАКЦИНА ДЛЯ СПЕЦИФИЧЕСКОЙ ПРОФИЛАКТИКИ ГРИППА(56) МЕЛЬНИКОВ С.Я. и др. Разработка и доклиническое исследование виросомальной расщепленной гриппозной вакцины нового поколения "Грифор". Журнал микробиологии,эпидемиологии и иммунобиологии, 2009,1, с. 21-26 Изобретение относится к области медицины и может быть использовано в вакцинопрофилактике. Предложена вакцина для профилактики гриппа, представляющая собой смесь поверхностных и внутренних антигенов вирусов гриппа типа А и В, полученная расщеплением очищенных вирионов детергентом октилгликозидом, в которой поверхностные антигены представлены в виде виросом размером 100-150 нм, включающих в свой состав собственные вирусные фосфолипиды,и что внутренние антигены представлены в виде мицелл, при соотношении гемагглютинина к внутренним антигенам 1:(0,5-2,5) и соотношении гемагглютинина к вирусным фосфолипидам 1:(2-4). Техническими результатами изобретения являются создание вакцины для профилактики гриппа с высокой иммуногенностью, протективной активностью и низкой реактогенностью, а также расширение арсенала противогриппозных вакцин. 016866 Изобретение относится к области медицины и может быть использовано в вакцинопрофилактике, в частности для профилактики гриппа. Уровень техники На протяжении последних десятилетий вакцины против гриппа постоянно совершенствовались с целью уменьшения реактогенности, но при этом зачастую снижалась иммуногенность, что заставляло разработчиков искать более совершенные формы презентации вирусных антигенов в составе вакцин или вводить в состав вакцин адъюванты. К настоящему времени для активной профилактики гриппа в основном применяют расщепленные("Ваксигрипп" (Санофи, Франция), "Флюарикс" (ГлаксоСмиткляйн, Бельгия) и виросомальные ("Инфлексал" (Берна, Швейцария), "Инвивак" (Солвей, Голландия вакцины. Однако существующие вакцины имеют ряд недостатков. Так, вакцины "Ваксигрипп" и "Флюарикс" обладают повышенной реактогенностью при сравнительно невысокой иммуногенности и индуцируют преимущественно гуморальный иммунитет. При этом они практически не индуцируют клеточный иммунитет, который очень важен для защиты организма от вируса гриппа. В состав вакцин "Инфлексал" и "Инвивак" не входят внутренние антигены (белок нуклеокапсида и мембранный белок), вследствие чего указанные вакцины также не способны активно индуцировать клеточный иммунитет. Кроме того, для формирования виросом в этих вакцинах применяют искусственный липид лецитин, что недостаточно технологично: количество образующихся виросом ниже по сравнению с использованием вирусных фосфолипидов, при этом качество упаковки гемагглютинина в виросомах,содержащих лецитин, существенно хуже. Наиболее близким аналогом вакцины в соответствии с настоящим изобретением является виросомальная расщепленная вакцина "Грифор", описанная в публикации Мельников С.Я. и др. Разработка и доклинические исследование виросомальной расщепленной гриппозной вакцины нового поколения "Грифор". ЖМЭИ. 2009.1, с. 21-26. Указанная вакцина обладает высокой антигенной и иммуногенной активностью и представляет собой смесь высокоочищенных протективных поверхностных и внутренних антигенов вирусов гриппа типа A и типа B. В публикации не приведено соотношение поверхностного антигена (гемагглютинина) к внутренним антигенам (белок нуклеокапсида и мембранный белок) и не обсуждается влияние этого соотношения на реактогенность препарата. Кроме того, в статье не раскрыто соотношение гемагглютинина и фосфолипидов в составе виросом. Размер виросом в описанной в статье вакцины не был определен с достаточной точностью. По приблизительным оценкам он находится в пределах от 90 до 180 нм. Таким образом, существует потребность в создании усовершенствованных вакцин для вакцинопрофилактики на основе подробного исследования влияния размера виросом, соотношения гемагглютинина к внутренним антигенам и соотношения гемагглютинина и вирусных фосфолипидов на реактогенность и иммуногенность препарата. Краткое описание фигур На фиг. 1 представлено микроскопическое изображение вакцины в соответствии с настоящим изобретением. На фиг. 2 представлена электрофореграмма полуфабрикатов (моновалентов) вакцины в соответствии с настоящим изобретением. Описание изобретения Авторы настоящего изобретения установили, что недостатки существующего уровня техники могут быть преодолены созданием вакцины, включающей собственные вирусные фосфолипиды, полученные в процессе расщепления вирионов. Было обнаружено, что применение собственных фосфолипидов вместо липида лецитина приводит к более эффективному и количественному образованию виросом. Было установлено, что оптимальный результат по антигенной активности на животных был получен с виросомальными структурами размером 100-150 нм. Виросомальные структуры меньшего размера(50-90 нм) обладали более низкой антигенной активностью, а структуры размером от 160 до 200 нм в существенной мере задерживались при стерилизующей фильтрации препарата и, таким образом, снижали выход лекарственной формы. Содержание фосфолипидов в предлагаемом препарате, превышающее отношение гемагглютинин:фосфолипиды 1:4, приводит к увеличению аггрегативности препарата и увеличению его реактогенности. Соотношение гемагглютинин:вирусные фосфолипиды менее 1:2 приводит к тому, что виросомальные структуры не образуются с высоким выходом, а упаковка гемагглютинина в них является неупорядоченной. В результате этого стабильность в процессе хранения препарата с таким соотношением гемагглютинин:фосфолипиды неудовлетворительно низкая. Было также установлено, что применение методики расщепления вирусов детергентом октилглюкозидом позволяет выделить внутренние антигены (белок нуклеокапсида и мембранный белок) в максимально нативном состоянии, что позволяет достигать повышения их иммуногенности по сравнению с известным уровнем техники. Было установлено, что если соотношение гемагглютинина и внутренних антигенов превышает 1:2,5, то полученный препарат обладает повышенной реактогенностью. Если соотношение гемагглютинин:внутренние антигены становится менее 1:0,5, то препарат индуцирует низкий-1 016866 уровень клеточного иммунитета у лабораторных животных и обладает неудовлетворительной протективной активностью. В соответствии с настоящим изобретением предлагается вакцина для профилактики гриппа, представляющая собой смесь поверхностных и внутренних антигенов вирусов гриппа типа A и B, полученная расщеплением очищенных вирионов детергентом октилгликозидом, в которой поверхностные антигены представлены в виде виросом размером 100-150 нм, включающих в свой состав собственные вирусные фосфолипиды, и внутренние антигены представлены в виде мицелл, при соотношении гемагглютинина к внутренним антигенам 1:(0,5-2,5) и соотношении гемагглютинина к вирусным фосфолипидам 1:(2-4), и имеющая коэффициент профилактической эффективности по меньшей мере 75% и индекс эффективности от 6,5 до 8,5. В результате проведенных исследований было установлено, что вакцина, характеризуемая вышеуказанными признаками, обладает более высокой иммуногенностью и протективной активностью, а также пониженной реактогенностью по сравнению с вакцинами известного уровня техники. Также создание вакцины в соответствии с настоящим изобретением расширяет арсенал противогриппозных вакцин за счет использования новых методов очистки и стабилизации антигенов. Технические результаты изобретения достигаются за счет применения разработанной авторами технологии расщепления и очистки антигенов, раскрытой в патенте РФ 2283139 (опубл. 10.09.2006). В результате применения этой технологии поверхностные антигены в составе предлагаемой вакцины представлены в виде виросом, а внутренние антигены - в виде мицелл с полным сохранением их антигенной нативности. Внутренние антигены вируса гриппа, выделенные с максимальным сохранением их молекулярной структуры, чрезвычайно важны для формирования противогриппозного клеточного иммунитета у вакцинируемых людей, что способствует усилению перекрестного иммунитета против дрейфующих штаммов вируса гриппа. Морфологический анализ методом электронной микроскопии (пример 1) показал, что в вакцине в соответствии с настоящим изобретением поверхностные антигены представлены в виде виросом размером 100-150 нм. Анализ состава полипептидов (пример 3) показал, что в вакцине соотношение гемагглютинина и внутренних антигенов находится в диапазоне 1:(0,5-2,5). Анализ состава фосфолипидов (пример 2) показал, что отношение гемагглютинина к фосфолипидам составляет 1:(2-4). Оценки иммуногенных и протективных свойств вакцины в соответствии с настоящим изобретением были получены в результате исследования реакции иммунной системы и организма в целом на экспериментальных животных, а также испытания вакцины на добровольцах. В результате исследований, описанных в примере 3, было установлено, что вакцина вызывает образование гемагглютинирующих антител у животных с незначительными колебаниями по различным серотипам вируса гриппа, что свидетельствует об эффективности предлагаемой вакцины. Проведенные доклинические исследования на животных (белых мышах, морских свинках, кроликах) позволили сделать следующие выводы: по показателям острой и хронической токсичности вакцина для лабораторных животных является нетоксичным и безвредным препаратом; в условиях хронического эксперимента на лабораторных животных выявлено, что вакцина хорошо переносится, не вызывает изменений в поведении, соматических и вегетативных реакций, а также не оказывает местно-раздражающего действия, т.е. обладает низкой реактогенностью. На основании I фазы клинического исследования на добровольцах (пример 5A) были сделаны следующие выводы. 1. Иммунизация предлагаемой вакциной не сопровождалась развитием нежелательных явлений в поствакцинальном периоде, а возникшие слабовыраженные общие и местные реакции были кратковременны и не вызывали ухудшения состояния здоровья привитых, что свидетельствовало о безопасности препарата и его низкой реактогенности. 2. Иммунизация добровольцев вакциной обеспечила следующие показатели иммуногенности к вирусу гриппа типов A(H1N1), A(H3N2) и B: уровень сероконверсии 55, 50 и 45%; уровень серопротекции 95, 90 и 100%; кратность прироста антител 3,8; 3,5 и 2,7 соответственно. Эти результаты свидетельствуют о высокой иммуногенности предлагаемой вакцины. На основании II фазы клинического исследования на добровольцах (пример 5 Б) были сделаны следующие выводы. 1. Вакцинация добровольцев предлагаемой вакциной в дозе 45 мкг и вакциной сравнения в дозе 45 мкг не сопровождалась развитием тяжелых местных и общих реакций в поствакцинальном периоде, а возникшие слабовыраженные реакции были кратковременны и не вызывали ухудшения состояния здоровья привитых, что свидетельствовало о безопасности препарата. 2. Однократная внутримышечная иммунизация добровольцев предлагаемой вакциной в дозе 45 мкг и вакциной сравнения ("Ваксигрип", Санофи Пастер, Франция) в дозе 45 мкг позволила обеспечить сле-2 016866 дующие показатели иммуногенности к вирусу гриппа серотипов H1N1, H3N2 и B. Предлагаемая вакцина в дозе 45 мкг обеспечивает: уровень сероконверсии 93,3, 88,4 и 77,4%; уровень серопротекции 90,6, 93 и 75,2%; кратность прироста антител 17,4; 10,0 и 6,8 соответственно. Вакцина сравнения в дозе 45 мкг обеспечивает: уровень сероконверсии 94,8, 92,2 и 82,3%; уровень серопротекции 96,1, 94,4 и 84,4%; кратность прироста антител 24,8; 11,0 и 11,5 соответственно. В результате изучения длительности сохранения поствакцинального иммунитета было показано,что уровень сохранения величины титров антител у добровольцев, привитых предлагаемой вакциной,был выше по отношению к титрам антител у добровольцев, привитых вакциной сравнения. Полученные результаты показали, что иммуногенность предлагаемой вакцины соответствует всем критериям, содержащимся в рекомендациях Комитета патентованных медицинских продуктов Евросоюза (Concept paper on the revision of the CPMP/BWP note for guidance on Note for guidance on harmonization Частота серопротекции = % с титрами анти-ГА выше 1:40. Частота сероконверсии = % с 4-кратным нарастанием титра анти-ГА после вакцинации в сравнении с титром до вакцинации, или % с титром анти-ГА после вакцинации выше 1:40 среди имевших до вакцинации титр анти-ГА менее 1:10. Фактор конверсии = средняя кратность увеличения СГТ (СГТпосле/СГТдо). Иммуногенность предлагаемой вакцины в отношении сохранения титров поствакцинальных антител выше, чем у вакцины сравнения. Результаты III фазы клинического исследования на добровольцах (пример 5 В) позволили рассчитать профилактическую эффективность предлагаемой вакцины, коэффициент эффективности которой в отношении лабораторно-подтвержденного гриппа составляет не ниже 75%, а индекс эффективности равен 6,5-8,5. Предпочтительно коэффициент эффективности равен 85-90%, а индекс эффективности 7,07,5. Высокие показатели профилактической эффективности предлагаемой вакцины по сравнению с известными зарубежными и отечественными вакцинами обусловлены несколькими причинами. Во-первых,в состав предлагаемой вакцины входят виросомы, которые способны увеличивать длительность поствакцинального иммунитета. Кроме того, в состав предлагаемой вакцины входят нативные мицеллы внутренних антигенов (мембранного белка и белка нуклеокапсида), которые помимо поверхностных антигенов способны индуцировать существенный уровень клеточного иммунитета. Наличие клеточного иммунитета существенно увеличивает защиту организма от вируса гриппа. Данное изобретение иллюстрируется следующими примерами, подтверждающими достижение его технических результатов. Для исследований использовали экспериментальные серии вакцины с содержание гемагглютинина серотипов A (H1N1 и H3N2) от 10(1,1) до 15(2,2) мкг, серотипа В - 152,2 мкг в одной прививочной дозе. Для конструирования вакцины использовали следующие вакцинные штаммы вируса гриппа: штаммB/Брисбен/60/08 (B), штамм А/Калифорния/7/2009 (H1N1), штамм A/Перт/16/09 (H3N2). Пример 1. Морфологические исследования полуфабрикатов (моновалентов) вакцины и готовой лекарственной формы вакцины. Морфологические изучения полуфабрикатов (моновалентов) вакцины и готовой лекарственной формы вакцины проводили методом электронной микроскопии с использованием технологии негативного контрастирования с использованием микроскопа Jeol 100C с увеличением 50000 раз. Для объективной оценки структурных особенностей вакцины был проведен сравнительный морфологический анализ следующих препаратов: 1) полуфабриката вакцины, серотипа A(H1N1); 2) полуфабриката вакцины, серотипа A(H3N2); 3) полуфабриката вакцины, серотипа B; 4) готовая лекарственная форма вакцины в соответствии с изобретением; 5) очищенного и концентрированного препарата вирионов вируса гриппа серотипа A(H1N1), из которого получен соответствующий полуфабрикат; 6) очищенного и концентрированного препарата вирионов вируса гриппа серотипа A(H3N2), из ко-3 016866 торого получен соответствующий полуфабрикат; 7) очищенного и концентрированного препарата вирионов вируса гриппа серотипа В, из которого получен соответствующий полуфабрикат. В процессе работы было исследовано 18 электронно-микроскопических сеточек, содержащих вышеперечисленные материалы, и просмотрено 500 полей зрения. Из указанного числа полей зрения были выбраны те, которые содержали структуры, наиболее типичные для данного препарата. При исследовании препаратов электронной микроскопией выявлены морфологические различия между вирионами вируса гриппа серотипов A(H1N1), A(H3N2) и B, с одной стороны, и вирусоспецифическими структурами - виросомами, содержавшимися в полуфабрикатах предлагаемой вакцины, полученных из соответствующих серотипов вируса гриппа, с другой. Содержавшиеся в полуфабрикатах предлагаемой вакцины виросомы характеризовались относительной мономорфностью и меньшими размерами по сравнению с вирионами соответствующих серотипов вируса гриппа. Виросомы имели преимущественно шаровидную форму и диаметр от 100 до 150 нм(фиг. 1). Гемагглютинины, выявляемые на поверхности виросом, имели четко выраженную морфологию,вертикальную ориентировку относительно липидной мембраны и достаточно плотную упаковку. В полуфабрикатах предлагаемой вакцины кроме виросом были обнаружены также мицелярные образования,состоящие из структурированного мембранного белка и из структур нуклеокапсида вируса гриппа. Пример 2. Определение количества фосфолипидов. Определение количества фосфолипидов в предлагаемой вакцине проводилось с помощью коммерческого диагностического набора фирмы Sentinel Diagnostics (Италия, каталожный номер 17320). Метод основан на том, что под действием фосфолипазы-D фосфолипиды гидролизуются до холина и фосфатидной кислоты. Затем холин окисляется холиноксидазой до бетаина и перекиси водорода, которая в присутствии пероксидазы реагирует с 4-аминоантипирином и производными фенола с образованием комплекса красного цвета. Оптическая плотность образующегося стабильного комплекса пропорциональна содержанию фосфолипидов в пробе. Калибровочные стандарты фосфолипидов (3 мг/мл) прилагаются в составе набора. Расчет количества фосфолипидов в составе предлагаемой вакцины проводился путем сравнения оптической плотности калибровочного стандарта и образцов вакцины. Согласно полученным результатам в различных полуфабрикатах вакцины (различные серотипы) отношение содержания гемагглютинина к содержанию фосфолипидов в 1 мл препарата существенно различалось. Наибольшее количество фосфолипидов (отношение 1:4) наблюдалось в полуфабрикатах серотипа В. В полуфабрикатах серотипов H1N1 и H3N2 это соотношение было в диапазоне 1:(2-3). Следует отметить, что это соотношение зависело также и от конкретной партии полуфабриката и зависело от технологии осаждения внутренних структур из полуфабриката. В конечном сведенном препарате вакцины соотношение гемагглютинина к фосфолипидам было в диапазоне 1:(2-4). Пример 3. Изучение состава полипептидов в полуфабрикатах вакцины и препаратах очищенных вирионов. Для сравнительного изучения состава полипептидов в полуфабрикатах вакцины и препаратах очищенных вирионов был использован метод электрофореза в 10% полиакриламидном геле с додецилсульфатом натрия в нередуцирующих условиях. Исследования проводились согласно МУК 4.1/4.2.558.96. Для определения чистоты, гомогенности и молекулярной массы генно-инженерных продуктов применяют метод вертикального электрофореза в полиакриламидном геле (ПААГ) с додецилсульфатом натрия (SDS) в нередуцирующих условиях с последующей окраской геля как Кумасси ярко-голубым R-250. Образование комплекса SDS с белком устраняет различия между белками, связанные с их зарядом, и переводит молекулы белков в конформацию, при которой радиус Стокса является функцией молекулярной массы белка. При соблюдении этих условий подвижность белков отражает их молекулярные массы. Полимеризацию геля ведут в ячейке, образованной стеклами и прокладками, определяющими размер и толщину геля. Сборку ячейки проводят в соответствии с инструкцией по работе с конкретным типом прибора для вертикального электрофореза. Описание метода проводится на примере аппарата "Протеан 11" (Био-Рад, США). Для данного прибора размеры стекол ячейки составляют: внешнее стекло 2220 см, внутреннее стекло - 2020 см. Размеры прокладок: длина - 20 см, ширина - 18 см, толщина 0,75-1,0 мм. При наличии приборов с другими характеристиками необходимо внести соответствующие коррективы в постановку испытаний. Для проведения электрофореза нижнюю камеру аппарата для электрофореза заполняют электродным буферным раствором и вставляют в нее электрофоретическую ячейку с гелем. Верхний резервуар заполняют электродным буферным раствором, удаляют из лунок пузырьки воздуха. Под слой буферного раствора в лунку вносят исследуемые образцы. Электрофорез проводят при температуре 10-14C в режиме постоянного тока. При прохождении фронта красителя через концентрирующий гель сила тока составляет 10-12,5 мА на 1 см 2 сечения геля (20-25 мА/гель). После вхождения фронта красителя в нижний разделяющий слой на 5-7 мм силу тока увеличивают до 20 мА на 1 см 2 сечения геля (40 мА/гель). После продвижения красителя на 14 см от нижнего края концентрирующего геля ток отключают, отделяют гель от стекол ячейки и переносят в кювету для окрашивания. Для того чтобы можно было выявить-4 016866 присутствие в исследуемом образце агрегатов и других компонентов, не входящих в гель, концентрирующий гель полностью не обрезают, оставляя 0,5-10 мм. В случае необходимости можно обрезать часть нижнего геля по фронту красителя. Нанесение образцов на гель проводят в следующем порядке. В лунки 1 и 10 вносят соответствующий (с бета-меркаптоэтанолом или без) буфер для приготовления образцов,разбавленный деионизованной водой в 4 раза. В лунки 2 и 9 вносят по 7 мкл смеси стандартных белков,например, фирмы "Фармация", Швеция. В остальные лунки вносят исследуемые образцы. При окрашивании Кумасси ярко-голубым R-250 вносят попарно такие объемы образца, чтобы количество белка в них составляло соответственно 10 и 40 мкг. При окрашивании геля нитратом серебра в зависимости от препарата в лунки вносят 2-5 мкг белка. Параллельно в одну из лунок вносят 0,002-0,01 мкг белка (в зависимости от чувствительности выявления минимальных количеств белка в данных условиях). Для окрашивания геля Кумасси ярко-голубым R-250 и его последующего обесцвечивания гель фиксируют 1 ч в фиксирующем растворе, после чего окрашивают в течение 16 ч. Затем окрашивающий раствор сливают, гель 2-3 раза споласкивают обесцвечивающим раствором и помещают в аппарат для обесцвечивания, заполненный обесцвечивающим раствором. Обесцвечивание проводят в течение 15 мин при напряжении 24 В. Гель вынимают из аппарата для обесцвечивания и споласкивают раствором для обесцвечивания и деионизованной водой. Сканирование обесцвеченного геля проводили с помощью аппаратаGel Dock 170-8170. В процессе анализа в одном и том же геле проводили сравнительный анализ полипептидов препаратов полуфабрикатов одного серотипа вируса гриппа, а также препаратов очищенных вирионов, из которых был получен соответствующий полуфабрикат. Эти результаты представлены на электрофореграмме (фиг. 2). Из представленных результатов следует, что в составе полуфабриката вакцины в соответствии с изобретением обнаруживаются те же полипептиды, что и в препарате очищенных вирионов, из которых был получен этот полуфабрикат. Но соотношение полипептида HA0 к полипептиду M1 в полуфабрикатах было значительно выше такового в препарате очищенных вирионов. Соотношение поверхностных и внутренних антигенов в предлагаемой вакцине меняется в зависимости от серотипа вируса, а также от технологических условий получения конкретной серии. Так в процессе получения полуфабриката очищенные вирионы разрушаются детергентом и поверхностные антигены, липиды вирусной мембраны, а также мембранный белок перешли в растворимое состояние. После удаления детергента и части вирусных липидов поверхностные антигены образовали псевдовирусные структуры виросомы, а мембранный белок образовал мицеллярные структуры различного размера. На конечной стадии технологического процесса, а именно стерилизующей фильтрации, часть наиболее крупных мицелл мембранного белка была задержана стерилизующей мембраной и, таким образом, не попала в состав полуфабриката вакцины. Таким образом, экспериментально установлено, что соотношение гемагглютинина и внутренних антигенов составляет 1:(0,5-2,5). Пример 4. Доклинические исследования на животных. Доклинические исследования безопасности предлагаемой вакцины в соответствии с настоящим изобретением проводят на основании методических рекомендаций Министерства здравоохранения и социального развития РФ "Доклинические испытания эффективности и безопасности новых иммунобиологических лекарственных препаратов" утверждены и введены в действие Приказом директора ФГБУ"ГИСК им. Л.А. Тарасевича" Минздравсоцразвития России от 31.12.201046-ОД. А) Оценка острой токсичности предлагаемой вакцины. Изучение острой токсичности вакцины в соответствии с изобретением было проведено на 40 мышах массой 18-20 г, рандомизированных на 2 группы - опытную и контрольную. Мышам контрольной группы однократно внутрибрюшинно вводили по 0,5 мл физиологического раствора, а опытной группе 10 мышам внутрибрюшинно вводили предлагаемую вакцину в дозе по 2250 мкг/кг (45 мкг/мышь) по гемагглютинину и 10 мышам - в дозе по 0,65 мкг/кг (0,013 мкг/мыши). Общая продолжительность наблюдения за животными при определении острой токсичности составила 7 дней. На 2-е сутки дислокацией шейных позвонков были умерщвлены по 4 мыши из опытной и контрольной групп, вскрыты и осмотрены. У всех этих мышей были забраны внутренние органы, взвешены и помещены в 10% формалин для дальнейших гистологических исследований. За мышами опытной и контрольной групп было продолжено наблюдение. Состояние мышей оценивали по внешнему виду, поведению, изменению массы тела и пищевой активности. За время наблюдения гибели белых мышей при однократном введении предлагаемой вакцины в дозе по 2250 мкг/кг и 0,65 мкг/кг не отмечено. На 8-е сутки 4-х мышей умерщвляли, проводили патологоанатомическую оценку внутренних органов и отбирали пробы крови. После взвешивания внутренних органов и определения их массы на момент забоя вычисляли коэффициенты масс (КМ) внутренних органов, после чего был проведен статистический анализ КМ и оценена достоверность полученных результатов с помощью критерия Стьюдента. За период наблюдения за группой животных гибели белых мышей не отмечено. У экспериментальных животных не изменялись поведение, двигательная активность, аппетит. Место нанесения препаратов у всех опытных мышей чистое, без воспаления и раздражения. Данные измерения массы тела мышей-5 016866 опытной группы на 2 и 8 сутки не отличалась от показателей массы тела животных контрольных групп больше чем на 5-8% (табл. 1). Таблица 1 Динамика изменения массы тела мышей после введения предлагаемой вакцины Анализ динамики результатов морфологических показателей крови при однократном введении предлагаемой вакцины в дозе 0,65 мкг/кг и 2250 мкг/кг не выявил изменений. Б) Оценка хронической токсичности предлагаемой вакцины. При оценке хронической токсичности в качестве контроля служили экспериментальные животные,которым в качестве плацебо вводили физиологический раствор (0,85% раствор натрия хлорида). Наблюдение за животными проводили ежедневно после введения препарата. Оценивали общее состояние, двигательную активность, поедание корма. Изучение хронической токсичности вакцины было проведено на 40 мышах массой 18-20 г, рандомизированных на 2 группы - опытную и контрольную. Мышам контрольной группы подкожно вводили по 0,2 мл физиологического раствора, а опытной группе ежедневно в течение 14 суток подкожно вводили по 0,2 мл вакцины в суммарной дозе 2250 мкг/кг по гемагглютинину (45 мкг/20 г мыши). Общая продолжительность наблюдения за животными при определении острой токсичности составила 15 дней. Внутрибрюшинное введение изучаемого препарата во всех дозах не вызывало гибели экспериментальных животных, во время наблюдения за животными не отмечено изменение двигательной активности, достоверно значимого изменения веса, аппетита, не было отмечено воспалительных явлений или раздражения слизистой. На 1-е сутки дислокацией шейных позвонков были умерщвлены по 4 мыши из опытной и контрольных групп, вскрыты и осмотрены. У всех этих мышей были забраны внутренние органы, взвешены и помещены в 10% формалин для дальнейших гистологических исследований. За мышами опытной и контрольной групп было продолжено наблюдение. На 15-е сутки 4 мышей умерщвляли, проводили патологоанатомическую оценку внутренних органов и отбирали пробы крови. После взвешивания внутренних органов и определения их массы на момент забоя вычисляли коэффициенты масс (КМ) внутренних органов, после чего был проведен статистический анализ КМ и оценена достоверность полученных результатов с помощью критерия Стьюдента(табл. 2). Таблица 2 Динамика изменения массы тела мышей после введения предлагаемой вакцины в течение 14 суток Следовательно, длительное подкожное введение предлагаемой вакцины не вызывало выраженного токсического эффекта у белых мышей за время наблюдения, это подтверждается данными морфологического и биохимического исследования крови. В) Оценка местно-раздражающего действия вакцины (мышиная модель). Местно-раздражающее действие вакцины оценивали на мышах в плантарном тесте путем введения-6 016866 вакцины в подушечку задней лапки. Опытной группе 1 вводили вакцину однократно в подушечку задней лапки в объеме 50 мкл (0,05 мкг/кг, 1/10 человеческой дозы). Опытной группе 2 вводили вакцину однократно в подушечку задней лапки в объеме 50 мкл (0,025 мкг/кг, 1/20 человеческой дозы). Неспецифическую воспалительную реакцию на введение вакцины оценивали с 24 ч и до 7 суток включительно. Индекс данной реакции определяли как процент прироста массы опытной лапки после введения вакцины по отношению к массе контрольной лапки, в которую вводили физиологический раствор (табл. 3). Таблица 3 Результаты по оценке местно-раздражающего действия вакцины в плантарном тесте Представленные данные свидетельствуют, что предлагаемая вакцина не вызывает местных воспалительных реакций на месте введения. Г) Оценка местно-раздражающего действия предлагаемой вакцины (кролики). Определение местно-раздражающего действия проводили на модели глаз кроликов по модифицированному методу Драйза. В группе по оценке местно-раздражающего действия вакцины использовали 3 кролика. Контролем служил левый глаз, в который капали по 50 мкл физиологического раствора, а в правый глаз на слизистую оболочку капали по 50 мкл изучаемого вакцинного препарата в дозе 45 мкг/2,5 кг кролика. Препарат наносили за веко кролика и придерживали веки закрытыми в течение 1 с. Реакцию регистрировали через 24, 48, 72 ч и на 7 сутки. Исследуемый материал оказывает раздражающее действие на глаза, если у 3 или более животных из опытной группы наблюдалась положительная реакция. При регистрации результатов конъюнктивальной пробы у 1-го кролика через 24 ч отмечалось расширение сосудов конъюнктивы, которое исчезало в течение последующих суток. У остальных кроликов применение предлагаемой вакцины не вызывало местного раздражающего действия. Д) Изучение антигенной активности предлагаемой вакцины. Готовят двукратные разведения сывороток в лунках плексигласовой доски, начиная с 1:10 до 1:640 и выше в объеме 0,2 мл. К каждому разведению сыворотки добавляют по 0,2 мл рабочей дозы антигена(4 АЕ). Смесь встряхивают, оставляют при температуре 202C на 30 мин, затем в каждую лунку добавляют по 0,4 мл 1%-ной суспензии куриных эритроцитов. Смесь повторно встряхивают, оставляют при температуре 202C в течение 40-45 мин (до оседания эритроцитов в контроле), после чего производят учет результатов реакции. При наличии специфических антител в сыворотке наступает задержка агглютинации эритроцитов. За титр сыворотки принимают предельное разведение, вызывающее полную задержку гемагглютинации. Задержка гемагглютинации указывает на соответствие типа антигена и взятой сыворотки; отсутствие задержки гемагглютинации свидетельствует о несоответствии типа взятой сыворотки. Препарат считают специфичным, если он не реагирует в РТГА с гетерологичной сывороткой. Для сравнительного изучения на мышах антигенной активности вакцин против гриппа шести беспородным белым мышам массой 18-20 г двукратно внутрибрюшинно с интервалом 14 суток вводили по 0,5 мл вакцины. Через 12-14 суток после повторной иммунизации полученные сыворотки мышей исследовали с гомологичными антигенами вакцины в РТГА согласно МУ 3.3.2.1758-03 "Методы определения показателей качества иммунобиологических препаратов для профилактики и диагностики гриппа", утв. 28 сентября 2003 г. Препарат должен вызывать нарастание антител в титрах не ниже 1:40 к каждому из трех входящих в ее состав штаммов вирусов гриппа типа А и В. В качестве контроля брали смесь сывороток от 2-3 неиммунных мышей. Результаты изучения антигенной активности предлагаемой вакцины и вакцины сравнения представлены в табл. 4.-7 016866 Таблица 4 Антигенная активность предлагаемой вакцины и вакцины сравнения против гриппа Полученные результаты свидетельствуют о том, что вакцина обладает более высокой антигенной активностью, чем вакцина сравнения. Пример 5. Клинические исследования на добровольцах. А) Первая фаза клинических исследований (изучение безопасности и иммуногенности на ограниченном контингенте добровольцев). Целью клинического исследования было изучение реактогенности, безопасности и иммуногенности препарата. В клиническом исследовании приняли участие 40 человек, в возрасте от 18 до 50 лет, полностью соответствующие критериям включения и исключения протокола клинических исследований. Методом случайной выборки все добровольцы были разделены на 2 группы. В 1 группе 20 человек были привиты предлагаемой вакциной с содержанием гемагглютинина (НА) - 45 мкг/доза - по 15 мкг НА штаммов вирусов гриппа серотипов H1N1, H3N2 и В. Во 2-й группе (контрольной) - 20 человек, в качестве плацебо получили раствор натрия хлорида изотонического в объеме 0,5 мл. Вакцину вводили однократно внутримышечно (в дельтовидную мышцу). Наблюдение за добровольцами проводили в течение 28 дней после вакцинации. При лабораторно-инструментальном исследовании общего и биохимического анализов крови, общего анализа мочи, общих IgM и IgA установлено, что все показатели были в пределах нормы. Вакцинация не сопровождалась развитием нежелательных явлений у добровольцев в поствакцинальном периоде,а возникшие слабовыраженные местные и общие реакции были кратковременны и не вызывали ухудшения состояния здоровья привитых, что свидетельствует о низкой реактогенности и безопасности вакцины. Иммуногенность предлагаемой вакцины (табл. 5) определяли в соответствии с указаниями, приведнными в МУ 3.3.2.1758-03, "Методы определения показателей качества иммунобиологических препаратов для профилактики и диагностики гриппа", утв. 28 сентября 2003 г. Таблица 5 Оценка иммуногенности предлагаемой вакцины ФС - фактор сероконверсии. СРМР - соответствие требованиям СРМР. На основании I фазы клинического исследования были сделаны следующие выводы. 1. Иммунизация предлагаемой вакциной не сопровождалась развитием нежелательных явлений в поствакцинальном периоде, а возникшие слабовыраженные общие и местные реакции были кратковременны и не вызывали ухудшения состояния здоровья привитых, что свидетельствовало о безопасности препарата. 2. К 28 дню поствакцинального периода наблюдалось снижение среднего уровня общегоIgE (Pcp0-280,001), что, вероятно, связано с входящими в состав вакцины специфическими структурами -8 016866 виросомами, способными изменять соотношение Thl/Th2 в сторону Th1. 3. Иммунизация добровольцев вакциной обеспечила следующие показатели иммуногенности к вирусу гриппа типов A(H1N1), A(H3N2) и B: уровень сероконверсии 55, 50 и 45%; уровень серопротекции 95, 90 и 100%; кратность прироста антител 3,8; 3,5 и 2,7 соответственно. У добровольцев с исходным титром антител, меньшим или равным 1:20, после вакцинации предлагаемой вакциной наблюдался 4-кратный прирост антител в 100% случаев к вирусу гриппа типа A (H1N1) и В и в 70% - к вирусу гриппа типа A (H3N2). Б) Вторая фаза клинических исследований (изучение безопасности и иммуногенности на расширенном контингенте добровольцев). В простом слепом сравнительном клиническом исследовании приняли участие 300 волонтеров, по 150 человек на каждой базе, полностью соответствующие критериям включения и исключения протокола. Методом случайного распределения все добровольцы были поделены на 2 группы. В 1 группе 50 человек были привиты вакциной с содержанием гемагглютинина 45 мкг/доза - по 15 мкг гемагглютинина штаммов вирусов гриппа серотипов H1N1, H3N2 и В. В 2 группе 50 человек были привиты препаратом сравнения (вакциной "Ваксигрип") с содержанием гемагглютинина - 45 мкг/доза - по 15 мкг гемагглютинина штаммов вирусов гриппа серотипов H1N1,H3N2 и В. Полученные при исследовании данные позволили сделать следующие выводы. 1. Вакцинация добровольцев предлагаемой вакциной в дозе 45 мкг и вакциной сравнения в дозе 45 мкг не сопровождалась развитием тяжелых местных и общих реакций в поствакцинальном периоде, а возникшие слабовыраженные реакции были кратковременны и не вызывали ухудшения состояния здоровья привитых, что свидетельствовало о безопасности препарата. 2. Однократная внутримышечная иммунизация добровольцев предлагаемой вакциной в дозе 45 мкг и вакциной сравнения в дозе 45 мкг позволила обеспечить следующие показатели иммуногенности к вирусу гриппа серотипов H1N1, H3N2 и В. Предлагаемая вакцина в дозе 45 мкг обеспечивает уровень сероконверсии 93,3%, 88,4% и 77,4%; уровень серопротекции 90,6%, 93% и 75,2%; кратность прироста антител 17,4; 10,0 и 6,8 соответственно. Вакцина сравнения в дозе 45 мкг обеспечивает уровень сероконверсии 94,8, 92,2 и 82,3; уровень серопротекции 96,1, 94,4 и 84,4%; кратность прироста антител 24,8; 11,0 и 11,5 соответственно. Полученные результаты показали, что иммуногенность предлагаемой вакцины, а также вакцины сравнения практически одинаковы и соответствуют всем критериям СРМР ЕМЕА. Согласно рекомендациям ВОЗ инактивированные гриппозные вакцины должны быть изучены на предмет длительности поствакцинального иммунитета на период 6 месяцев, т.е. 180 дней. В результате изучения длительности сохранения поствакцинального иммунитета было показано, что уровень сохранения величины титров антител у добровольцев, привитых предлагаемой вакциной, был выше по отношению к титрам антител у добровольцев, привитых вакциной сравнения (табл. 6).-9 016866 Таблица 6 Оценка длительности поствакцинального иммунитета В) Третья фаза клинических исследований (изучение профилактической эффективности). Профилактическую эффективность вакцины оценивали по двум показателям: индексу эффективности и коэффициенту эффективности в соответствии с санитарными правилами СП 3.3.2.561-96. Для изучения профилактической эффективности предлагаемой вакцины в многоцентровом, контролируемом, проспективном, рандомизированном исследовании приняли участие около 6000 добровольцев(мужчины и женщины) в возрасте от 18 лет и старше, рандомизированные в две группы. Основная группа добровольцев (группа вакцинированных) была привита предлагаемой вакциной в дозе 45 мкг, группе наблюдения вакцину не вводили. В соответствии с рекомендациями заболевание "грипп" диагностировали на основании клинических данных. Для подтверждения клинического диагноза "грипп" среди вакцинированных и невакцинированных участников исследования проведены лабораторно-серологические исследования парных сывороток в первые дни заболевания и через 14-21 день, а также оценены клиническое течение и тяжесть заболевания. Общая продолжительность наблюдения для каждого участника клинического исследования составила не менее 6 месяцев. Проведенный в сформированных группах добровольцев анализ заболеваемости гриппом в эпидемический сезон 2007-2008 гг. позволил рассчитать, что профилактическая эффективность предлагаемой вакцины составляет по меньшей мере 79,5% при индексе эффективности, равном по меньшей мере 6,8. Профилактическую эффективность вакцины оценивали по двум показателям: индекс эффективности: K=b/a,где K - индекс эффективности;b - заболеваемость среди невакцинированных. В случае заболевания и лабораторного подтверждения диагноза "грипп" у добровольцев была сопоставлена заболеваемость, оценены клиническое течение и тяжесть заболевания, возникновение осложнений. В основной и контрольной группах добровольцев на всем протяжении исследования проводили оценку заболеваемости и тяжести клинического течения гриппа, а также осуществляли дифференциальную диагностику гриппа с другими вирусными и бактериальными инфекциями. Грипп диагностировали на основании клинических данных, а именно выявления основных симптомов: высокая температура,достигающая 39-40C, длительностью около 3-4 дней; озноб; насморк; першение и боль в горле; сухой,"лающий" кашель, который сохранялся в течение 2 недель; общая выраженная слабость, недомогание,разбитость; боль и "ломота" в мышцах и суставах; одышка; боль при движении глаз; конъюнктивит. Кроме того, для подтверждения клинического диагноза грипп среди вакцинированных и невакцинированных участников исследования проведены лабораторно-серологические исследования парных сывороток в первые дни заболевания и через 14-21 день, а также оценены клиническое течение и тяжесть заболевания. Общая продолжительность наблюдения для каждого участника клинического исследования составила не менее 6 месяцев.- 10016866 Проведенный анализ заболеваемости гриппом и ОРВИ в эпидемический сезон 2007-2008 гг. у привитых добровольцев и наблюдательной группы (невакцинированные) в Санкт-Петербурге, Томске и Перми позволил рассчитать профилактическую эффективность предлагаемой вакцины, коэффициент эффективности которой в отношении лабораторно-подтвержденного гриппа составляет 86,5%, а индекс эффективности 7,4. Ранее в России в рамках государственных испытаний проводилась оценка профилактической эффективности различных зарубежных вакцин против гриппа. В результате этого исследования было показано, что для вакцины "Инфлювак" (Солвей Фарма, Голландия) индекс эффективности составил 4,5, а коэффициент эффективности - 77,7%. Для вакцины "Ваксигрип" (Санофи Пастер, Франция) коэффициент эффективности вакцины составил 79%, а индекс эффективности составил 4,7. Вакцина "Грипол" (Петровакс, Россия) является наиболее применяемой отечественной вакциной. В ходе государственных испытаний было установлено, что данная вакцина имеет коэффициент эпидэффективности 59,3%; а индекс эпидэффективности равен 2,0. Приведенные выше результаты исследований подтверждают более высокие показатели профилактической эффективности предлагаемой вакцины по сравнению с известными зарубежными и отечественными вакцинами. ФОРМУЛА ИЗОБРЕТЕНИЯ Вакцина для профилактики гриппа, представляющая собой смесь поверхностных и внутренних антигенов вирусов гриппа типа А и В, полученная расщеплением очищенных вирионов детергентом октилгликозидом, в которой поверхностные антигены представлены в виде виросом размером 100-150 нм,включающих в свой состав собственные вирусные фосфолипиды, и внутренние антигены представлены в виде мицелл, причем соотношение гемагглютинина к внутренним антигенам составляет 1:(0,5-2,5), а к вирусным фосфолипидам составляет 1:(2-4).
МПК / Метки
МПК: A61K 39/145, A61P 31/16
Метки: профилактики, гриппа, специфической, вакцина
Код ссылки
<a href="https://eas.patents.su/13-16866-vakcina-dlya-specificheskojj-profilaktiki-grippa.html" rel="bookmark" title="База патентов Евразийского Союза">Вакцина для специфической профилактики гриппа</a>
Рекомбинантная псевдоаденовирусная наночастица, фармацевтическая композиция для профилактики или терапии гриппа (варианты), способ профилактики или терапии гриппа

Номер патента: 16517
Опубликовано: 30.05.2012
Авторы: Тутыхина Ирина Леонидовна, Саакян Седа Аветиковна, Шмаров Максим Михайлович, Народицкий Борис Савельевич, Седова Елена Сергеевна, Иванова Татьяна Ильинична, Логунов Денис Юрьевич, Грибова Ирина Юрьевна, Гинцбург Александр Леонидович, Рутовская Марина Владимировна, Васильев Лев Анатольевич, Тиллиб Сергей Владимирович
МПК: A61P 31/16, C07K 16/10, A61K 39/145...
Метки: композиция, наночастица, рекомбинантная, способ, псевдоаденовирусная, гриппа, варианты, фармацевтическая, терапии, профилактики
Формула / Реферат:
1. Рекомбинантная псевдоаденовирусная наночастица, несущая экспрессирующую кассету, содержащую нуклеотидную последовательность, которая кодирует однодоменное мини-антитело, связывающееся с вирусом гриппа типа А или типа В.2. Псевдоаденовирусная наночастица по п.1, причем вирус гриппа типа А характеризуется антигенной формулой H1N(1-9).3. Псевдоаденовирусная наночастица по п.1, причем вирус гриппа типа А характеризуется антигенной формулой...
Вакцина для иммунизации позвоночных против бруцеллеза, вакцина для профилактики или лечения позвоночных от бруцеллеза и способ получения вакцинного штамма, аттенюированный или авирулентный вариант штамма бактерий в. abortus rb51, способ профилактики или лечения позвоночных от бруцеллеза

Номер патента: 5234
Опубликовано: 30.12.2004
Авторы: Бойл Стивен М., Срирнаганатан Наммалвар, Корбил Лайнетт, Шуриг Герхард Г., Краверо Сильвио, Вемулапалли Рамеш
МПК: C12N 15/00, A61P 37/02, A61K 39/00...
Метки: аттенюированный, rb51, штамма, вакцина, abortus, против, вакцинного, бактерий, получения, вариант, иммунизации, бруцеллеза, позвоночных, лечения, профилактики, авирулентный, способ
Формула / Реферат:
1. Вакцина для иммунизации позвоночных против бруцеллеза, отличающаяся тем, что она содержит аттенюированный или авирулентный штамм бактерий рода Brucella, который способен к сверхэкспрессии по меньшей мере одного гомологичного антигена, кодируемого по меньшей мере одним геном бактерий рода Brucella, причем указанный антиген способен индуцировать защитный иммунный ответ против бруцеллеза. 2. Вакцина по п.1, отличающаяся тем, что аттенюированный...
Вакцина для профилактики эхинококкоза животных

Номер патента: 6309
Опубликовано: 27.10.2005
Авторы: Ахметсадыков Нурлан Нуралдинович, Уразбекова Дурия Сембековна, Муминов Абдулло, Шодмонов Иброим Шодмонович
МПК: A61P 33/10, A61K 39/00
Метки: животных, вакцина, эхинококкоза, профилактики
Формула / Реферат:
Вакцина для профилактики эхинококкоза животных, включающая эффективное количество ферментативно расщепленных сколексов, жидкости и герментативной оболочки эхинококка с последующим гидролизом, отличающаяся тем, что в ее состав кроме сколексов входят иммуноадъювант геля (гидроокись алюминия) и формалин.
Композиция (варианты) и способ лечения или профилактики инфекции гриппа у млекопитающего

Номер патента: 3989
Опубликовано: 25.12.2003
Авторы: Бишофбергер Норберт У., Лью Уиллард, Хичкок Майкл Дж.М., Ким Чанг Ю., Лиу Хонгтао, Дал Теренс К., Милз Роджер Г., Уильямс Метью Э.
МПК: A61K 31/215, A61K 31/33, A61K 31/396...
Метки: варианты, композиция, способ, лечения, млекопитающего, гриппа, профилактики, инфекции
Формула / Реферат:
1. Композиция для лечения или профилактики инфекции гриппа, содержащая эффективное количество соединения общей формулы где E1 означает -CO2R5, -CO2R5aW5 или -CO2W5; G1 означает -N(R5)2, -N(R11)C(N(R11))(N(R11)2) или -C(R11)2-N(R11)2; T1 означает -NH(C(O)CH2F) или -NH(C(O)CHF2); U1 означает -SR4, -NHR4 или -N(R4)2; каждый R1 независимо означает водород или алкил, содержащий от 1 до 12 атомов углерода; каждый R2 независимо означает R3 или R4,...
Композиция для получения реассортантного рекомбинантного вируса гриппа, способ получения указанного вируса и реассортантный рекомбинантный вирус гриппа

Номер патента: 12965
Опубликовано: 26.02.2010
Автор: Каваока Йошихиро
МПК: C12N 7/04, C12N 15/86, C12N 7/02...
Метки: получения, вируса, композиция, указанного, рекомбинантный, рекомбинантного, реассортантный, вирус, гриппа, способ, реассортантного
Формула / Реферат:
1. Композиция для получения "6+2" реассортантного рекомбинантного вируса гриппа с высоким титром, содержащая множество векторов вирусов гриппа, включающая:а) векторы для продуцирования вРНК, включающие вектор, содержащий промотор, функционально связанный с кДНК белка РА вируса гриппа, соединенной с последовательностью терминации транскрипции; вектор, содержащий промотор, функционально связанный с кДНК белка РВ1 вируса гриппа,...
Предыдущий патент: Способ обработки волос
Следующий патент: Амортизатор транспортного средства
Случайный патент: Высотное здание