Гетероциклические соединения для обезболивания и их применение.












Формула / Реферат
1. Соединение формулы (I)

где R1 является Н, С1-6 алкилом или С6-12 арилом, который может быть замещен группами -СООН, -NH2 или гуанидином;
R2 и R3 являются, независимо друг от друга, Н, ОН, C1-6 алкилом, -C(NH)-NH2, положительно заряженной группой или С7-13 аралкилом, который может быть замещен NH2, ОН, C1-6 алкилом или галогеном; или
R2 и R3 совместно образуют 5- или 6-членное кольцо, которое может содержать гетероатом;
R4 является Н, C1-6 алкилом, OR6, SR6 или N(R6)2, где каждый R6 является, независимо, Н, С1-3 алкилом или галогеном;
Х является О, S, SO, SO2, N-R5, где каждый R5 является, независимо, Н, C1-6 алкилом или С7-13 аралкилом, который может быть прерван одним или более гетероатомом(атомами);
n является целым числом от 0 до 2;
m является целым числом от 0 до 3.
2. Соединение по п.1, отличающееся тем, что Х выбран из S и SO2, m равно 3 и n равно 0.
3 Соединение по п.2, отличающееся тем, что Х является S.
4 Соединение по п.3, отличающееся тем, что R2 является Н и R3 выбран из Н, ОН и -C(NH)-NH2.
5 Соединение по п.3, отличающееся тем, что R1 является метилом.
6 Соединение по п.3, отличающееся тем, что R4 выбран из ОН и метокси.
7 Соединение по п.6, отличающееся тем, что R4 является ОН.
8 Соединение по п.1, выбранное из
#8b 5,6,7,8,9,11,12-гептагидро-3-метокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амина,
#9 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амина (сульфазоцин),
#10 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-гидроксиламина,
#8а 5,6,7,8,9,11,12-гептагидро-3-метокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-гидроксиламина,
#9а (-)-транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина,
#9b (-)-транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина,
#11 транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-гуанидина,
#12 транс-5,6,7,8,9,11,12-гептагидро-10-сульфоно-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина
9. Соединение по п.1, выбранное из
#9 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амина (сульфазоцин), и
#9a (-)-транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина.
10. Способ обезболивания млекопитающего путем введения ему фармацевтически эффективного количества соединения формулы (I) по п.1.
11. Способ по п.10, отличающийся тем, что Х выбран из S и SO2, m равно 3 и n равно 0.
12. Способ по п.11, отличающийся тем, что Х является S.
13. Способ по п.10, отличающийся тем, что R2 является Н и R3 выбран из Н, ОН и -C(NH)-NH2.
14. Способ по п.10, отличающийся тем, что R1 является метилом и R4 является ОН.
15. Способ по п.10, отличающийся тем, что вышеупомянутое соединение выбрано из:
#8b 5,6,7,8,9,11,12-гептагидро-3-метокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амина;
#9 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амина (сульфазоцин);
#10 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-гидроксиламина;
#8а 5,6,7,8,9,11,12-гептагидро-3-метокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-гидроксиламина;
#9а (-)-транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина;
#9b (+)-транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина;
#11 транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-гуанидина;
#12 транс-5,6,7,8,9,11,12-гептагидро-10-сульфоно-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина.
16. Способ активации опиоидных рецепторов млекопитающего путем введения ему соединения формулы (I) по п.1, активирующего опиоидный рецептор.
17. Способ по п.16, отличающийся тем, что Х выбран из S и SО2, m равно 3 и n равно 0.
18. Способ по п.16, отличающийся тем, что R2 является Н и R3 выбран из Н, ОН и -C(NH)-NH2.
19. Способ по п.16, отличающийся тем, что R1 является метилом и R4 является ОН.
20. Способ по п.16, отличающийся тем, что вышеупомянутое соединение выбрано из
#8b 5,6,7,8,9,11,12-гептагидро-3-метокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амина,
#9 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амина (сульфазоцин),
#10 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-гидроксиламина,
#8a 5,6,7,8,9,11,12-гептагидро-3-метокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-гидроксиламина,
#9а (-)-транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина,
#9b (+)-транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина,
#11 транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-гуанидина,
#12 транс-5,6,7,8,9,11,12-гептагидро-10-сульфоно-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина.
21. Применение фармацевтически эффективного количества соединения формулы I по п.1 в качестве обезболивающего средства для млекопитающего.
22. Применение соединения формулы I по п.1 в качестве средства, активирующего опиоидный рецептор млекопитающего.
Текст
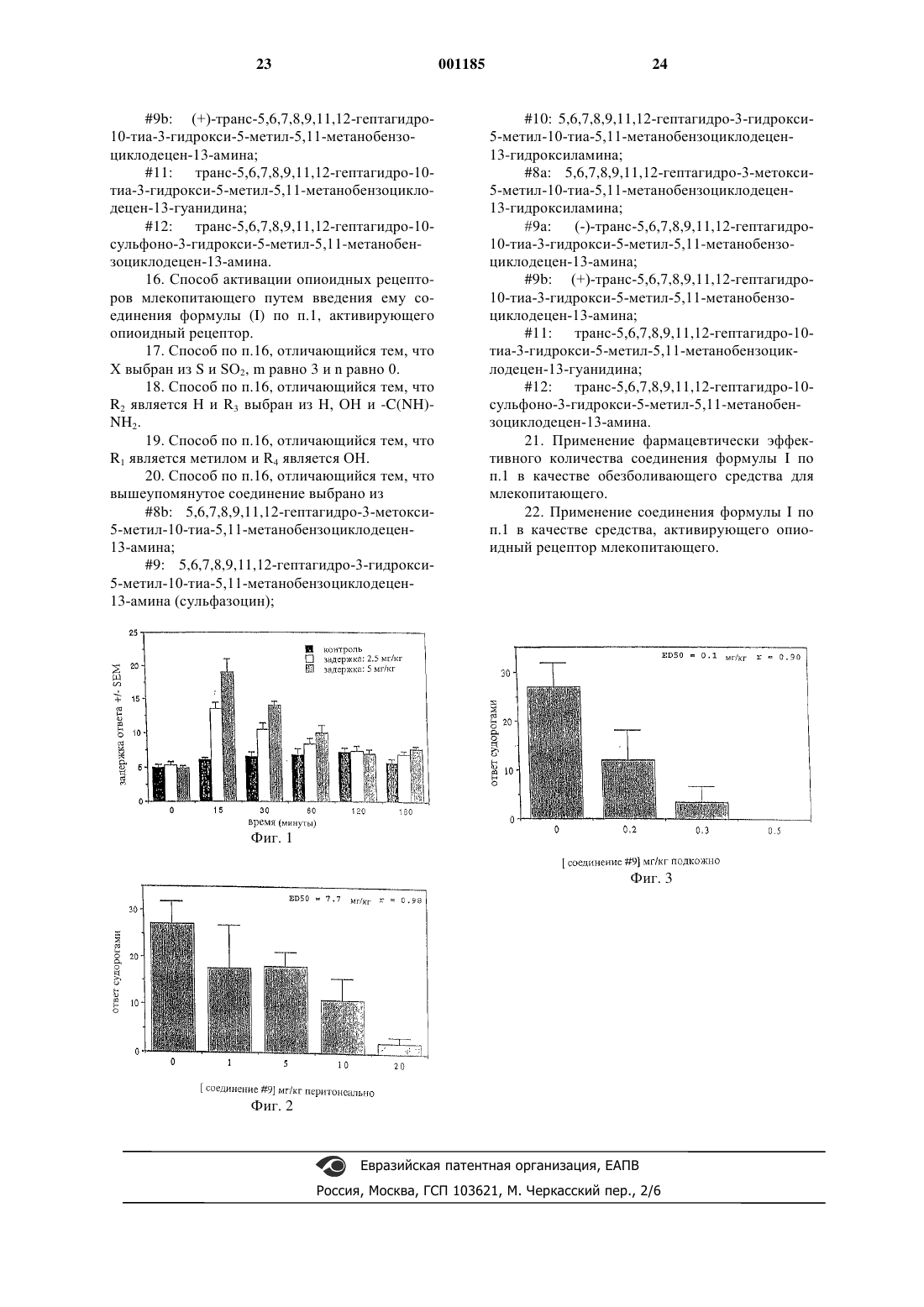
1 Область изобретения Настоящее изобретение касается новых полициклических агонистов опиоидных рецепторов, обладающих обезболивающим действием, и их приемлемых фармацевтических составов. В другом аспекте изобретение относится к способам и применениям, имеющим отношение к новым агонистам и составам. Предпосылки изобретения Наркотические опиоидные анальгетики остаются основой доступных сейчас лекарственных средств для облегчения от умеренной до серьезной боли. Опиоидные обезболивающие средства обеспечивают характерный антиболевой эффект у различных видов животных(включая homo sapiens) путем активации специфических рецепторов в центральной нервной системе. Согласно современным моделям оценки боли на животных является хорошо обоснованным, что активация одного или нескольких этих рецепторов обеспечивает антиболевые эффекты. Показано, что у высших животных существует множество типов опиоидных рецепторов,среди которых были охарактеризованы, по крайней мере, три различных класса с доказательствами существования дополнительных классов или подклассов: мю , каппаи дельта . Например, см. В. Мартин с соавторами (W. Martin et al., J. Pharmacol. Exp. Ther.,197, p. 517 (1975); и Дж. Лорда с соавторами (J.-Рецептор локализован в головном мозгу и оказывается вовлеченным в анальгетический эффект морфиноподобных лекарств. Активация-рецептора в головном и спинном мозге оказывается способной обеспечивать обезболивание, особенно на спинномозговом уровне. Рецептор найден в некоторых периферических тканях в дополнение к головному и спинному мозгу и проявляет дифференцированное сродство к эндогенным опиоидным белкам, известным как энкефалины. Наконец, хотя это и сомнительно, что -рецепторы являются строго"опиоидными" по своей природе, т.к. они активируются неопиоидными соединениями, большая часть психотомиметических эффектов опиоидных лекарств, такие как дисфории и галлюцинации, оказываются опосредованы этим классом рецепторов. Сущность изобретения Настоящее изобретение обеспечивает соединения, имеющие обезболивающее действие,которые являются новым полициклическим агонистом опиоидных рецепторов, имеющими общую структуру, представленную формулой I формула I где R1 является Н, С 1-6 алкилом или С 6-12 арилом, выборочно замененными на полярные группы;R2 и R3 являются независимо Н, ОН, C1-6 алкилом, -C(NH)-NH2, положительно заряженной группой или C7-13 аралкилом, выборочно замещенными на NH2, ОН, С 1-6 алкил или галоген; или R2 и R3 вместе образуют 5- или 6 членное кольцо, выборочно содержащее гетероатом;N(R6)2, отличающиеся тем, что каждый R6 является независимо Н, C1-3 алкилом или галогеном; Х является О, S, SO, SO2, N-R5 или C-(R5)2,отличающиеся тем, что каждый R5 является независимо Н, C1-6 алкилом или C7-13 аралкилом,выборочно прерванными одним или более гетероатомами;n является целым числом от 0 до 2;m является целым числом от 0 до 3. В другом аспекте настоящего изобретения обеспечивается способ агонистики опиоидных рецепторов млекопитающего, состоящий из введения вышеупомянутому млекопитающему соединения, представленного формулой (I) в количестве, агонистичном опиоидному рецептору. В другом аспекте обеспечивается способ стимулирования обезболивания у млекопитающего, состоящий из введения вышеупомянутому млекопитающему фармацевтически эффективного количества соединения, представленного формулой (I). Специалисту в данной области очевидно,что соединения формулы (I) в зависимости от заместителей могут содержать один или более хиральных центров и таким образом существовать в форме многих различных изомеров, оптических изомеров (т.е. энантиомеров) и их смесей, включая рацемические смеси. Все такие изомеры, энантиомеры и их смеси, включая рацемические смеси, включены в рамки изобретения. Изобретение также обеспечивает фармацевтически приемлемые составы, состоящие из соединений формулы (I), для использования в устранении боли. Изобретение также обеспечивает фармацевтически приемлемые составы, содержащие соединения формулы (I), для использования как помощь при диагностике и/или как исследовательские инструменты, такие как меченые лиганды, радиоактивные проводники для позитронной эмиссионной томографии или как па 3 рамагнитные агенты для ЯМР процессов, опосредованных опиатными рецепторами. Далее, изобретение обеспечивает использование соединения формулы (I) для производства терапевтических средств для обезболивания. Кроме того, изобретение обеспечивает использование соединения формулы (I) для производства химических соединений для использования в качестве помощи при диагностике и/или как исследовательские инструменты, такие как меченые лиганды, радиоактивные проводники для позитронной эмиссионной томографии или как парамагнитные агенты для ЯМР процессов,опосредованных опиатным рецептором. Краткое описание чертежей Фиг. 1 показывает дозозависимое ингибирование соединением 9, введенным мышам подкожно, задержки ответа на теплоту излучения; фиг. 2 - дозозависимое ингибирование соединением 9, введенным мышам перорально,ответа судорогами (PBQ); фиг. 3 - дозозависимое ингибирование соединением 9, введенным мышам подкожно,ответа судорогами (PBQ). Детальное описание изобретения Следующие общепринятые сокращения использованы в заявке и в формуле изобретения. Термин "ED50", как показано в табл. 1 дляPBQ анализа судорог, определен как доза лекарства, которая вызывает 50% снижение количества наблюдаемых судорог по сравнению с контрольным опытом. Термин "ED50", используемый в анализе с горячей пластиной (фиг. 1) определен как доза лекарства, требуемого для двукратного увеличения задержки ответа по сравнению с контрольным опытом, и был определен логарифмическим пробит-анализом. Термин "Ki" - это константа ингибирования связывания. Термин "Кi/Кi" является величиной, которая может быть использована для измерения селективности. Это отношение представляет собой соотношение аффинностей соединений для связывания с - и -рецепторами. Будучи использованным в этой заявке,термин "алкил" представляет насыщенный или ненасыщенный, замещенный (на галоген, гидроксил, амино или С 6-20 арил) или незамещенный; прямую цепь, разветвленную цепь или циклическую углеводородную долю, отличающиеся тем, что вышеупомянутые прямая цепь,разветвленная цепь или циклическая углеводородная доля могут быть прерваны одним или больше гетероатомами (такими как кислород,азот или сера). Термин "гетероатом" как используется в дальнейшем представляет собой N, O и S, также как SO и SO2. 4 Термин "арил" представляет собой карбоциклическое ядро, которое может быть замещено (например, C1-6 алкилом, галогеном, гидроксилом, аминогруппой), прервано, по крайней мере, одним гетероатомом (например, N, О илиS) и может содержать, по крайней мере, одно кольцо бензоидного типа (например, фенил и нафтил). Термин "аралкил" представляет группу арилов, присоединенную к соседнему с алкилом атому (например, бензил). Соединения настоящего изобретения представлены формулой (I), как определено выше. Предпочтительно R1 является циклогексилом. Предпочтительно R1 является фенилом,произвольно замещенным полярными группами. Предпочтительно полярными группами являются СООН, NH2 или гуанидин. Более предпочтительно R1 является Н. Наиболее предпочтительно R1 является СН 3. Предпочтительно R2 является Н. Предпочтительно R3 является ОН. Наиболее предпочтительно R3 является Н. Предпочтительно R4 является ОСН 3. Предпочтительно R4 является ОН. Предпочтительно Х является NH. Более предпочтительно Х является О. Наиболее предпочтительно Х является S. Предпочтительно R5 является C1-6 алкилом. Более предпочтительно R5 является СН 3. Наиболее предпочтительно R5 является Н. Предпочтительно n является 0. Предпочтительно m является 3. Предпочтительное соединение изобретения включает: Соединение 8b: 5,6,7,8,9,11,12-гептагидро-3-метокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амин. Предпочтительное соединение изобретения включает: Соединение 9: 5,6,7,8,9,11,12-гептагидро 3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амин (сульфазоцин). Предпочтительное соединение изобретения включает: Соединение 10: 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-гидроксиламин. Предпочтительное соединение изобретения включает: Соединение 9 а: (-)-транс-5,6,7,8,9,11,12 гептагидро-10-тиа-3-гидрокси-5-метил-5,11 метанобензоциклодецен-13-амин; Предпочтительное соединение изобретения включает: Соединение 11: транс-5,6,7,8,9,11,12 гептагидро-10-тиа-3-гидрокси-5-метил-5,11 метанобензоциклодецен-13-гуанидин Предпочтительное соединение изобретения включает: 5 Соединение 12: транс-5,6,7,8,9,11,12 гептагидро-10-сульфоно-3-гидрокси-5-метил 5,11-метанобензоцикло-децен-13-амин; Более предпочтительное соединение изобретения включает: Соединение 9: 5,6,7,8,9,11,12-гептагидро 3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амин (сульфазоцин); и Соединение 10: 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-гидроксиламин; Наиболее предпочтительное соединение изобретения включает: Соединение 9: 5,6,7,8,9,11,12-гептагидро 3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амин (сульфазоцин); и Соединение 9 а: (-)-транс-5,6,7,8,9,11,12 гептагидро-10-тиа-3-гидрокси-5-метил-5,11 метанобензоциклодецен-13-амин; Предпочтительные соединения настоящего изобретения могут быть получены с использованием традиционных методов синтеза и способов выделения, известных специалистам в области органического и биоорганического синтеза, которые обеспечивают новые и уникальные комбинации для полного синтеза каждого соединения. Предпочтительные схемы получения промежуточных соединений, вовлеченных в синтез, как и конечных соединений настоящего изобретения, следуют ниже. Успешный синтез этих соединений возможен посредством различных схем синтеза, одна из которых представлена на схеме 1. Схема 1 Этапы, проиллюстрированные на схеме 1,могут быть кратко описаны следующим образом: Шаг 1: Соединение I, алкил-1-тетралон обрабатывали подходящим реактивом Гриньяра типа метил магнийбромида в сухом неполярном растворителе, таком как ТГФ (тетрагидрофуран),для получения третичного спирта - соединения 6 Шаг 2: Спирт (соединение II) дегидратировали в кислой среде, такой как насыщенный водныйNH4Cl, с получением соединения III. Шаг 3: Двойную связь в позиции 1 на олефине эпоксидировали с использованием стандартных реагентов и растворителей, таких как магниевая соль монопероксифталевой кислоты в изопропаноле, с образованием эпоксида, соединениеIV. Шаг 4: Эпоксид подвергали перегруппировке в кислой среде, такой как водный растворNаНСО 3, с использованием стандартных методов для получения кетона, соединение V. Шаг 5: Алкилирование бис-алкил-2-тетрона (соединение V) проводили в щелочных условиях в неполярном растворителе с использованием реактива дигалогеналкила, типа дибромбутана,с получением соединения VI. Шаг 6: Нуклеофильное замещение бромида выполняли подходящим ацилирующим агентом,таким как тиоацетат калия, для получения соединения VII. Шаг 7: Позицию 3 ацилированного тетралона (соединение VII) галогенизировали в неполярном растворителе, типа смеси бензола и сухого ТГФ,используя соответствующий реагент и неполярный растворитель, такой как бромин в сухом ТГФ, для получения соединения VIII. Шаг 8: Боковую цепь циклизовали в щелочной среде с использованием стандартных реагентов и растворителей, как, например, бромид лития и сухой ТГФ под аргоном, с добавлением основания типа метоксида натрия, получая полициклическое соединение (соединение IX). Шаг 9:Keтогруппу соединения IX преобразовывали в алкилоксим, используя стандартные процедуры, хорошо известные в данной области, с образованием соединения X. Шаги 10 и 11: Соединение Х восстанавливали с использованием комплекса Боран-ТГФ. Если реакцию проводили в тетрагидрофуране, то получали смесь 50:50 соединений XI и XII. Если реакцию проводили в диглиме (2-метоксиэтиловый эфир), то селективно получали амин (соединение ХII). Шаг 12: Соединение XI может быть рециклизовано и восстановлено до амина с использованием комплекса Боран-ТГФ в диглиме с получением соединения XII. Принимается во внимание, что соединения настоящего изобретения могут быть модифицированы cпециалистами в данной области таким 7 образом, как присоединение меток, таких как радиоактивные метки, что позволяет предложить соединения для использования в качестве радиопроводников. Соединения настоящего изобретения могут быть использованы как агонисты опиатного рецептора in vitro или ex vivo,как в случае радиомеченых агентов, радиопроводников для использования в позитронэмиссионной томографии, парамагнитных агентов для использования в изображении магнитного резонанса и в случае антагонистов кальциевого канала, связанного с NMDAрецептором. Принимается во внимание, что соединения настоящего изобретения могут быть модифицированы специалистами в данной области таким образом, как предотвращение доступа в центральную нервную систему, таким образом, что они могут функционировать как агонисты опиатного рецептора в периферических тканях. Настоящее изобретение также обеспечивает фармацевтические составы, которые содержат фармацевтически эффективное количество соединения этого изобретения или их фармацевтически приемлемые соли и, выборочно,фармацевтически приемлемые носитель или адъювант. Термин "фармацевтически эффективное количество" относится к количеству соединения, требуемому для введения млекопитающему для индуцирования обезболивания. Также термин "агонистичное количество опиоидного рецептора" относится к такому количеству соединения, введенного млекопитающему,которое необходимо для связывания и/или активирования опиоидных рецепторов in vivo. Терапевтические способы этого изобретения состоят из стадий лечения пациентов фармацевтически приемлемым способом этими соединениями или составами. Такие составы могут быть в форме таблеток, капсул, капсулированных таблеток, порошков, гранул, леденцов,суппозиториев, общих порошков или жидких препаратов, таких как оральные или стерильные парентеральные растворы или суспензии. Терапевтические агенты настоящего изобретения могут быть введены по отдельности или в комбинации с фармацевтически приемлемыми носителями. Пропорция каждого носителя определяется растворимостью и химической природой соединения путем введения и стандартной фармацевтической практикой. Для получения консистенции для введения предпочтительно, чтобы состав изобретения находился в форме единичной дозы. Формы,представляющие единичную дозу для орального введения, могут являться таблетками и капсулами и могут содержать традиционные эксципиенты. Например, связывающие агенты, такие как акация, желатин, сорбитол или поливинилпирролидон; наполнители, такие как лактоза,сахар, кукурузный крахмал, фосфат кальция, 001185 8 сорбитол или глицин; таблетирующие смазки,такие как стеарат магния; дезинтеграторы, такие как крахмал, поливинилпирролидон, крахмал бриколлата натрия или микрокристаллическая целлюлоза; или фармацевтически приемлемые увлажняющие агенты, такие как лаурилсульфат натрия. Соединения могут быть введены парентерально; это означает внутримышечно, внутривенно или подкожно. Для парентерального введения могут быть использованы соединения в форме стерильных растворов, содержащих другие растворы, например, солевой раствор или глюкозу, достаточных, чтобы сделать раствор изотоническим. Соединения могут быть введены орально в форме таблеток, капсул или гранул, содержащих подходящие экципиенты, такие как крахмал, лактоза, белый сахар и подобное. Соединения могут быть введены орально в форме растворов, которые могут содержать агенты, придающие цвет и/или запах. Соединения могут быть также введены подъязычно в виде трахеи или леденцов, в которых каждый активный ингредиент смешан с сахарным или кукурузным сиропами, агентами, придающими запах, и красителями, и затем достаточно дегидратированы,чтобы сделать смесь пригодной для прессования в твердую форму. Твердые оральные составы могут быть приготовлены традиционнымиспособами смешивания, наполнения, таблетирования или подобными. Повторные операции смешивания могут быть использованы для распределения активного агента в тех композициях, где используются большие количества наполнителей. Такие процедуры являются, конечно, традиционными в области. Таблетки могут быть покрыты в соответствии с хорошо известными в нормальной фармацевтической практике способами, особенно оболочками, растворяющимися в кишечнике. Оральные жидкие препараты могут быть в форме эмульсий, сиропов или эликсиров, или могут быть представлены как сухие продукты,воспроизводимые при помощи воды или других подходящих растворителей перед использованием. Такие жидкие препараты могут содержать или не содержать традиционные добавки. Например, суспендирующие агенты, такие как сорбитол, сироп, метилцеллюлозу, желатин,гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или гидрированные съедобные жиры; эмульгирующие агенты,такие как моноолеат сорбитана или акацию; неводные растворители (которые могут включать съедобные масла), такие как миндальное масло, фракционированное кокосовое масло,масляные эфиры, выбранные из группы, состоящей из глицерина, пропиленгликоля, этиленгликоля и этилового спирта; предохраняю 9 щие агенты, например, метилпарагидроксибензоат, этилпарагидроксибензоат, n-пропилпарагидроксибензоат или n-бутилпарагидроксибензоат сорбитоловой кислоты; и, если требуется, традиционные агенты, придающие запах или цвет. Для парентерального введения могут быть приготовлены формы жидкой единичной дозы путем использования соединения и стерильного растворителя, и, в зависимости от используемой концентрации, могут быть либо суспендированы, либо растворены в растворителе. Оказавшись в растворе, соединение может быть впрыснуто и простерилизовано фильтрацией перед наполнением подходящего пузырька или ампулы с последующим запечатыванием переносчика или упаковки для хранения. Вспомогательные средства, такие как местные анестетики, предохраняющие или буферные агенты, могут быть растворены в переносчике перед использованием. Стабильность фармацевтических композиций может быть усилена путем замораживания композиции после наполнения пузырька и удаления воды под вакуумом (например,высушиванием с замораживанием состава). Парентеральные суспензии могут быть приготовлены, по существу, тем же образом, за тем исключением, что соединение должно быть скорее суспендировано в переносчике, чем растворено,и далее стерилизация не достигается фильтрацией. Соединение может быть стерилизовано,однако, путем обработки его окисью этилена перед суспендированием в стерильном пузырьке. Для обеспечения однородного распределения соединения в его состав могут быть внесены поверхностно-активные вещества или смачивающие вещества. Фармацевтические составы этого изобретения состоят из фармацевтически эффективного количества соединения этого изобретения и фармацевтически приемлемого носителя. Обычно они содержат от примерно 0,1% до примерно 99% по весу, предпочтительно от примерно 10% до примерно 60% по весу соединения этого изобретения в зависимости от используемого способа введения. Настоящее изобретение также обеспечивает способ обезболивания таких пациентов, как млекопитающие, включая человека, который состоит из введения пациенту фармацевтически эффективного количества соединения, его фармацевтически приемлемой соли или фармацевтического состава, как описано выше. Врачи определят такую дозировку настоящих терапевтических агентов, которая будет наиболее подходящей. Дозировки могут варьировать в зависимости от способа введения и выбора определенного соединения. Дозировка дополнительно может варьировать при лечении определенного пациента. Дозировка соединения, использованного в лечении, будет варьировать в зависимости от степени расстройства,веса пациента, относительной эффективности 10 соединения и выбора лечащего врача. Такая терапия может продлиться в течение нескольких недель, с перерывами или без перерывов, до момента, когда симптомы пациента устранены. Были получены и исследованы в качестве агонистов опиоидных рецепторов гетероциклические соединения общей формулы I. Эти соединения представлены в табл. 1 вместе с их соответствующими константами связывания,ингибирования и ингибиторной активностью вPBQ анализе. Эти результаты показывают, что соединения изобретения являются эффективными как обезболивающие агенты. Соединения настоящего изобретения могут быть использованы как агонисты опиатных рецепторов in vitro или ex vivo, как в случае,например, радиомеченых агентов, радиопроводников, парамагнитных агентов. Соединения изобретения могут быть полезны как в исследовательских целях, так и в диагностике. Для получения соединений этого изобретения могут быть использованы различные способы в зависимости от исходных и/или промежуточных продуктов. Успешное получение этих соединений достигается путем нескольких возможных направлений синтеза, некоторые из которых показаны ниже. Для дальнейшей помощи в понимании настоящего изобретения приведены следующие примеры таких соединений-агонистов опиатного рецептора. Эти примеры, конечно, не должны толковаться как лимитирующие настоящее изобретение. Их вариации, известные в настоящее время или развитые позже, которые могут находиться в компетенции специалиста в данной области, рассматриваются как подпадающие в рамки настоящего изобретения, как описано в настоящем документе. Пример 1. Получение 1,2-дигидро-7 метоксиметилнафталина. 7-Метокси-1-тетралон (25 г) высушивали азеотропной перегонкой с толуолом и растворяли в сухом ТГФ (200 мл). Раствор охлаждали до-70 С (под аргоном) и добавляли бромид метилмагния (1,4 М в смеси толуол/ТГФ, 187,5 мл). Объединенную реакционную смесь оставляли для перемешивания при комнатной температуре в течение ночи. Смесь осторожно обрабатывали водным насыщенным растворомNН 4 Сl и экстрагировали этилацетатом. Последний раствор промывали соленой водой, высушивали над MgSO4 и выпаривали. Остаток растворяли в бензоле (150 мл), добавляли p-TSOH 11 и смесь нагревали с обратной посадкой ДинаСтарка до окончания реакции дегидратации. Этот раствор бензола разводили этилацетатом,промывали раствором NaHCО 3, высушивали над MgSО 4 и выпаривали. Остаток экстрагировали гексаном, пропускали через колонку с силикагелем и элюировали смесью гексана и этилацетата (1:0, 400:1, 200:1). Выход продукта 20,87 г (84,42%). 1 Н ЯМР (300 МГц, CDCl3) : 2.05 (s, 3 Н); 2.23 (m, 2H); 2.69 (t, 2H); 3.81 (s, 3H); 5.88 (m,1H); 6.68-7.06 (m, 3 Н) частей на миллион. Пример 2. Получение 7-метокси-1-метил 2-тетралона.(20,87 г) растворяли в изопропаноле (100 мл) и охлаждали в ледяной бане. Добавляли магниевую соль монопероксифталиевой кислоты(mmpp) (17 г), затем добавляли воду (50 мл) и перемешивали смесь при комнатной температуре в течение 2 ч. Когда окисление было закончено, реакционную смесь гидролизовали водным раствором NаНСО 3, частично выпаривали и экстрагировали этилацетатом. Последний экстракт промывали соляным раствором и выпаривали. Остаток растворяли в смеси этанола (156 мл), воды (121 мл) и конц. H2SО 4 (24,3 мл) и кипятили в атмосфере N2 в течение 3 ч, охлаждали и нейтрализовали раствором NaHCO3. После частичного выпаривания остаток экстрагировали этилацетатом, промывали солевым раствором, высушивали над MgSО 4 и выпаривали. Продукт очищали на колонке с силикагелем,используя смесь гексана и этилацетата (100:1,50:1, 50:1,5). Выход продукта составил 16,2 г 7-Мeтокси-1-метил-2-тетралон (4 г) высушивали азеотропной перегонкой с толуолом,растворяли в сухом ТГФ (THF) (150 мл), охлаждали в ледяной бане в атмосфере аргона, добавляли раствор натрия-бис-(триметилсилил)амида(NaHMDS) (1 M в ТГФ, 23,13 мл) и перемешивали в течение 1/2 ч. Добавляли 1,4 дибромобутан (9,78 мл), оставляли реакционную смесь для нагревания до комнатной темпе 001185 12 ратуры на ночь, после чего ее гидролизовали солевым раствором, экстрагировали этилацетатом, высушивали над MgSО 4 и выпаривали. Смесь продукта очищали на колонке с силикагелем, используя смесь гексана и этилацетата (200:1, 150:1, 100:1, 75:1 и 50:1). Выход продукта составил 5,19 г (76%). 1 1-(4'-бромобутил)-1-метил-7-метокси-2 тетралон (4,48 г) высушивали азеотропной перегонкой с толуолом и растворяли в сухом DMF(25 мл). Добавляли триацетат калия (5,86 г) и оставляли смесь для перемешивания в атмосфере аргона на ночь. Смесь экстрагировали этилацетатом, промывали солевым раствором, высушивали над MgSО 4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь гексана и этилацетата (75:1, 50:1, 20:1). Выход продукта составил 3,9 г (88,3%). 1 Н ЯМР (300 МГц, СDСl3) : 0.99 (m, 2H); 1.5 (m, 6H); 2.07 (m, 1H); 2.25 (s, 3H); 2.65 (m,4H); 2.95 (m, 2H); 3.79 (s, 3H); 6.71-7.08 (m, 3H) частей на миллион. Пример 5. Получение эпимерной смеси 3 бромо-1-(4'-ацетотиобутил)-1-метил-7-метокси 2-тетралона. 1-(4'-ацетотиобутил)-1-метил-7-метокси-2 тетралон (4 г) высушивали азеотропной дистилляцией толуола. Его растворяли в смеси бензола(244 мл) и сухого ТГФ (64 мл) и перемешивали при комнатной температуре под атмосферой аргона. Бромид (0,8 мл) растворяли в сухом ТГФ (26 мл) и постепенно добавляли к реакционной смеси в атмосфере аргона. После 1 ч перемешивания смесь продукта гидролизовали водным раствором NаНСО 3, экстрагировали этилацетатом, промывали солевым раствором,высушивали над MgSO4 и выпаривали. Остаток высушивали азеотропной дистилляцией в толуоле и далее высушивали под высоким вакуумом. Пример 6. Получение 5,6,7,8,9,11,12 гептагидро-3-метокси-5-метил-10-тиа-5,11 этанобензоциклодецен-13-она.(примерно 6,25 ммоль) высушивали азеотропной дистилляцией толуола и растворяли в сухом ТГФ (200 мл), добавляли бромид лития (сухой,0,54 г), дегазировали раствор аргоном при комнатной температуре в течение одного часа и охлаждали в ледяной бане, хорошо перемешивая с пропусканием через смесь аргона. Растворяли метоксид натрия (0,5 М в метаноле, 13,75 мл) в сухом ТГФ (75 мл), дегазировали аргоном при комнатной температуре в течение одного часа, после чего спринциванием добавляли к последнему раствору в течение 4 ч. Объединенную реакционную смесь перемешивали еще в течение 1/2 ч, разбавляли этилацетатом (100 мл), промывали солевым раствором, высушивали над MgSО 4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь гексана и этилацетата (75:1, 50:1). Выход продукта составил приблизительно 50-55%. Он кристаллизовался при стоянии. 1 Н ЯМР (300 MГц, CDCl3) : 1.4-1.95 (m,4H); 2.85 (m, 2 Н); 2.7-3 (m. 2H); 3.4 (m, 1H); 3.82(5,2 мл). Объединенную смесь нагревали до 80 С в течение двух дней. Ее охлаждали, разбавляли CH2Cl2 и промывали солевым раствором. После высушивания над MgSO4 растворитель выпаривали, остаток очищали на колонке с силикагелем, используя смесь гексана и этилацетата (50:1, 25:1, 10:1, 5:1, 2:1). Выход продукта составил 1,22 г (92%). 1 Н ЯМР (300 МГц, CDCl3) : 1.2-1,9 (m,9H); 2.4 (m, 2H); 2.85 (m, 2H); 3.2 (dd, 1H); 3.8 (s,3H); 5.11 (t, 1H); 6.6-7.1 (m, 3H) частей на миллион. Высушивали 5,6,7,8,9,11,12-гептагидро-3 метокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-оксим (смесь изомеров, 0,3 г) с толуолом и растворяли в сухом ТГФ (30 мл). Его охлаждали в ледяной бане в атмосфере аргона. Добавляли комплекс боран-ТГФ (1 М раствор в ТГФ, 7,87 мл) и объединенную смесь кипятили в течение 30 ч, охлаждали в ледяной бане. Осторожно добавляли воду (0,4 мл) и концентрированную НСl (0,6 мл) в соответствующем порядке. Cмесь кипятили в течение 15 мин, охлаждали и высушивали. Остаток подщелачивали концентрированным NH4OH до рН 12, экстрагировали СН 2 Сl2, промывали солевым раствором, высушивали над MgSO4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь гексанa и этилацетата (50:1, 20:1,10:1, 5:1, 5:1,5, 2:1, 1:1 и 1:2). Выход 5,6,7,8,9,11,12-гептагидро-3-метокси-5-метил 10-тиа-5,11-метанобензоциклодецен-13-гидроксиламина составил 0,073 г (23,1%). Он кристаллизовался из смеси этилацетата и гексана. 1 Н ЯМР (300 МГц, СDCl3) : 1.1-1.91 (m,9H); 2.3 (m, 2H); 3.30 (d, 1H); 3.37 (m, 2H); 3.7IR (пленка): 1612, 3300 см-1 Масс-спектрометрия: 293.8, 275.8, 260.8. Структура соединения 8 а была подтверждена рентгеновской кристаллографией выборочного кристалла. Выход 5,6,7,8,9,11,12-гептагидро-3 метокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амина составил 0,0968 г (32%). Свободное основание было растворено в гексане. 1 Н ЯМР (300 МГц, CDCl3) : 0.8-2.5 (m,11H); 3.18 (m, 3H); 3.6 (q, 1H); 3.8 (s, 3H); 6.6-7.1(m, 3H) частей на миллион. Этот продукт растворяли в эфире (40 мл) и подкисляли смесью метанол-HCl. Позволяли суспензии осаждаться и фильтровали. Осадок промывали эфиром и высушивали, получая 0,090 г продукта. 1 Н ЯМР (300 МГц, CDCl3) : 0.8-1.7 (m,6H); 1.8 (m, 2H); 2.0-2.5 (m, 3H); 3.45 (m, 2H); 3.5 (m, 2H); 3.8 (S, 3H) 6.7-7.1 (m, 3H) ppm. Масс-спектрометрия, m/z: 278. Пример 9. Получение 5,6,7,8,9,11,12 гептагидро-3-гидрокси-5-метил-10-тиа-5,11 метанобензоциклодецен-13-амина (сульфазоцина) соединение 9. Высушивали 5,6,7,8,9,11,12-гептагидро-3 метокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амин (0,260 г) азеотропной перегонкой с толуолом и растворяли в сухом СН 2 Сl2 (40 мл). Охлаждали до -70C в атмосфере аргона. Добавляли раствор трибромида бора (1 М раствор в СН 2 Сl2, 187 мл) и при температуре окружающей среды оставляли объединенную смесь перемешиваться на ночь. Реакционную смесь гидролизовали раствором NаНСО 3, понижали рН при помощи раствора NH4OH до 12 и экстрагировали СН 2 Сl2. Последний раствор высушивали над MgSO4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь толуола и этилацетата (10:1, 5:1, 2:1, 1:1,1:2). Выход 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-амина составил 0,162 г (65%). 1H ЯМР (350 МГц, DMSO-D6) : 1.02 (m,1H); 1.25 (m, 5H); 1.55 (m, 2H); 2.01 (m, 2H); 2.55 (m, 1H); 2.97 (d, 1H) 3.08 (m, 1H), 3.14 (m,2H); 6.4-6.9 (m, 3H) частей на миллион. Вышеупомянутый продукт был преобразован в его хлористо-водородную соль и очищен с помощью HPLC (жидкостной хроматографией под высоким давлением). Пример 10. Получение 5,6,7,8,9,11,12 гептагидро-3-гидрокси-5-метил-10-тиа-5,11 метанобензоциклодецен-13-гидроксиламина соединение 10. Растворяли 5,6,7,8,9,11,12-гептагидро-3 метокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-гидроксиламин (0,166 г) в смеси уксусной кислоты (4,5 мл) и 48% НВr (4,5 мл). Полученную смесь охлаждали, осторожно нейтрализовали NаНСО 3 и экстрагировали СН 2Cl2. Последний раствор промывали солевым раствором, высушивали над MgSO4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь гексана и этилацетата (10:1,10:1,5, 5:1). Выход 5,6,7,8,9,11,12-гептагидро-3 гидрокси-5-метил-10-тиа-5,11-метанобензоциклодецен-13-гидроксиламина составил 0,035 г(m, 1H); 6.6-7.0 (m, 3H) частей на миллион. Вышеупомянутый продукт был преобразован в его хлористо-водородную соль и очищен с помощью НРLС (жидкостной хроматографией под высоким давлением). Пример 11. Получение соединения 9 а (-)транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидро 16 кси-5-метил-5,11-метанобензоциклодецен-13 амина. Соединение 9 (2,96 г) смешивали с Dтартроновой кислотой (2,01 г), растворенной в кипящем этаноле (95%, 50 мл), и фильтровали. Нерастворимую массу промывали горячим этанолом (25 мл). Объединенные фильтраты выпаривали досуха и осадок вновь растворяли в горячем этаноле (20 мл). Полученную пушистую твердую массу вновь растворяли в горячем этаноле (15 мл) и оставляли без движения при комнатной температуре в течение 2 дней для кристаллизации. Полузакристаллизованную массу далее еще дважды подвергали подобной операции фракционной кристаллизации. Было обнаружено, что проба на этой стадии имеет диастерометрическую чистоту 98% согласно хиральной производной peareнта Марфи (0,267 г). Тартроновую соль названного соединения(0,1 г) растворяли в горячем метаноле (20 мл) и переносили на колонку с ионообменной смолойAmberlite IRA-400 (Сl- форма) (5 г, последовательно промытую метанолом, водой, 0,1 М НСl,водой и метанолом). Колонку последовательно промывали метанолом (100 мл) и водой (100 мл). Объединенные элюенты выпаривали и лиофилизовали. Остаток (0,076 г). Пример 12. Получение соединения 9b (+)транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13 амина. Соединение 9-D-тартроновую соль (высокообогащенную декстровращающимся диастереомером 1,5 г) смешивали с NH4OH (10 мл),насыщенным хлоридом натрия, и экстрагировали метиленхлоридом. Последний промывали солевым раствором, высушивали над MgSО 4 и выпаривали. Остаток (1,27 г) смешивали с Lтартроновой кислотой (0,87 г) и кипятили с этанолом (95%, 100 мл), фильтровали, затем фильтрат оставляли кристаллизоваться при комнатной температуре в течение 2 дней. Осажденную массу оставляли медленно кристаллизоваться из горячего изо-пропанола. Было обнаружено, что проба имеет диастереометрическую чистоту 97% согласно хиральной производной реагента Марфи, с выходом 0,1515 г. Тартроновую соль названного соединения(0,076 г) растворяли в горячем метаноле (25 мл) и переносили на колонку, набитую активированным Amberlite IRA-400 (Сl-форма, 5 г), которую затем последовательно промывали метанолом (100 мл) и водой (100 мл). Объединенные фильтраты выпаривали и лиофилизировали,остаток (0,058 г). Пример 13. Получение соединения 11 транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13 гуанидина. Соединение 9 (0,35 г) высушивали азеотропной перегонкой с толуолом и растворяли в сухом пиридине (5 мл). Добавляли 1 Н-пиразол 1-карбоксамидин гидрохлорид (1,29 г) и диизопропилэтиламин (1,74 мл). Объединенную смесь нагревали до 80 С в атмосфере азота в течение 4 дней. Растворитель выпаривали и очищали остаток на колонке с силикагелем, используя смесь метиленхлорида и метанола. Продукт (0,41 г) растворяли в метаноле, насыщенном хлористым водородом (5 мл) и выпаривали растворитель. Остаток очищали HPLC(жидкостной хроматографией под высоким давлением), с выходом 0,045 г конечного продукта. Пример 14. Получение соединения 12 транс-5,6,7,8,9,11,12-гептагидро-10-сульфо-3 гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина. Соединение 9 (0,1 г) высушивали азеотропной перегонкой с толуолом, растворяли в сухом метиленхлориде (20 мл) и охлаждали в ледяной бане в атмосфере аргона. Добавляли трифторуксусный ангидрид (0,54 мл) и пиридин(0,5 мл). После перемешивания при комнатной температуре в течение ночи реакционную смесь гидролизовали водным раствором бикарбоната натрия, экстрагировали метиленхлоридом, промывали солевым раствором, высушивали надMgSО 4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь гексана и метиленхлорида. Выход 0,068 г транс 5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5 метил-5,11-метанобензоциклодецен-13 трифторацетамида. Транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3 гидрокси-5-метил-5,11-метанобензоциклодецен 13-трифторацетамида растворяли в смеси этанола (2 мл) и воды (1 мл) и затем охлаждали в ледяной бане. Добавляли монопероксифталевую кислоту, гексагидратную соль магния (0,21 г). Через 1 ч добавляли насыщенный водный раствор бикарбоната натрия (5 мл). Объединенную смесь перемешивали при комнатной температуре в течение ночи и выпаривали, остаток экстрагировали метиленхлоридом. Полученный раствор промывали солевым раствором и выпаривали, получая 0,123 г транс-5,6,7,8,9,11,12 гептагидро-10-сульфо-3-гидрокси-5-метил-5,11 метанобензоциклодецен-13-трифторацетамида. 18 Транс-5,6,7,8,9,11,12-гептагидро-10 сульфо-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-трифторацетамид (0,123 г) высушивали азеотропной перегонкой с толуолом и добавляли безводный гидразин (2 мл). Смесь перемешивали при комнатной температуре в течение 2 дней, затем выпаривали досуха под вакуумом. Полученный продукт растворяли в метаноле (2 мл) и оставляли при комнатной температуре. Кристаллическое вещество (0,037 г) отфильтровывали, растворяли в насыщенном растворе хлористого водорода в метаноле (2 мл) и выпаривали. Остаток лиофилизовали, получая 0,031 г названного соединения. Исследования активности Обезболивающую активность соединений изобретения определяли in vivo на PBQ модели судорог и тесте на горячей пластине на грызунах. Ингибирование индуцированных PBQ (фенилбензохиноном) судорог у мышей является оценкой как центрально, так и периферически опосредованного обезболивания. Эксперименты проведены Sigmund et al., Proc. Soc. Ex. Biol.Med., 95, стр. 729 (1957), приведенного здесь в качестве ссылки. Центрально опосредованное обезболивание определяли ингибированием ответа на горячей пластине у мышей. Эксперименты проведены согласно G.Woolfe и A. Macdonald, J. Pharmacl. Exp.Ther.,80, стр. 300 (1994), приведенного здесь в качестве ссылки. Опыты по измерению аффинностей связывания опиоидного рецептора для ,ирецепторов, как и GPI и MDV анализы, были проведены согласно Schiller et al., Biophys. Res.Commun., 85, p. 1322 (1975); Rothman et al., Peptides, Vol. 11, pp. 311-331, 1990; Kaffa et al., Peptides 15 (3), 401-404, 1994; Fowler et al., Neurochem. Int 24(5), 401-426, 1994; и Leslie F., Pharmacological Review 39(3), 197-249, 1987, приведенных здесь в качестве ссылки. Пример 15. Анализ связывания радиорецептора. А. Приготовление мембраны. Крыс Sprague-Dawley мужского пола весом между 350-450 г подвергали ингаляции СО 2. Крысы были обезглавлены. Minus cerebellum мозга удалили и поместили в ледяной солевой раствор, затем гомогенизировали в ледяном 50MM трис-буфере рН 7,4 (10 мл/мозг). Мембраны центрифугировали при 14000 rpm в течение 30 мин при 4 С. Шарики ресуспендировали,примерно 6 мл/мозг в 50 мМ ледяном трисбуфере, рН 7,4, и хранили при -78 С до готовности к использованию. Подсчет уровня белков гомогената мозга проводили в соответствии с набором для анализа белков, приобретенном уBio-Rad. Б. Анализ радиолиганда. В качестве радиолигандов дляирецепторов были использованы (H)-DAMGO (Н)DADLE, соответственно, 50 л радиолиганда, 19 100 л мембран и серийно разведенное исследуемое соединение инкубировали в течение 1 ч при 22 С. Определяли неспецифическое связывание,используя 500-кратный избыток немеченого лиганда в присутствии проводников и мембран. Свободный лиганд отделяли от связанного фильтрацией через бумагу Whatman GF/B 20 ледяным Трисом, рН 7,4, используя сборник клеток Brandelа. Фильтры высушивали и подсчитывали радиоактивность в микроплате на 24 лунки в присутствии 500 мл сцинтиллята на лунку. Радиоактивность измеряли, используя счетчик Wallac 1450 Microbeta. Кривые замещения рисовали, используя программу Microsoft Excel. Определяли Ki для различных соединений по значениям IС 50 в соответствии с уравнением Cheng и Prusoff. Таблица Связывание рецептора и ингибиторная активность in vivo исследуемых соединений по PBQ-анализу (мыши)nM 9 3,70,45 711 nm 143 0,2 7,6 0,1 10 61,5 10 uM 0,2 9 а 0,68 41 0,093 9b 200 3257 16,37 11 35,8 1 Химическим раздражителем, использованным для индуцирования ответа судорогами у мышей, являлся фенилбензохинон. ED50 определяли спустя 20 мин после введения лекарства. Пример 16. Анализ судорог на фенилхинон. А. Подопытные. Тест проводили, используя CD1 мышей мужского пола (Charles River) весом от 19 до 25 г. Животных содержали при постоянных параметрах света, температуры и влажности. Животные проходили акклиматизацию в течение трех дней перед экспериментом. Б. Приготовление лекарства и процедура дозировки. Раствор фенилхинона (0,02%) готовили следующим образом. 20 мг фенилхинона растворяли в 5 мл 90% этанола. При постоянном перемешивании и медленном нагревании добавляли раствор фенилхинона к 95 мл дистиллированной воды. Раствор фенилхинона защищали от света все время и каждый день готовили новый раствор для теста. Рекомендуется ждать 2 ч перед использованием раствора фенилхинона. Все исследуемые соединения растворяли в дистиллированной воде и вводили подкожно или орально. Мышей инъецировали внутриперитонеально раствором 0,02% фенилхинона (2-фенил 1,4-бензохинон, Sigma). Фенилхинон инъецировали в различные интервалы времени - 20, 60,120 и 180 мин после введения соединения (или растворителя, или стандарта). Пример 17. Анализ на горячей пластине на мышах. А. Подопытные. Для этого теста использовали CD1 мышей мужского пола (Charles River) весом от 20 до 25 г. Мышей взвешивали, идентифицировали и произвольно распределяли по группам из 10. Б. Приготовление лекарства и процедура введения. Соединения (или стандарт, или растворитель) обычно вводили мышам подкожной инъекцией или орально. С. Подсчет обезболивающей активности. Мышей оценивали индивидуально на предмет времени задержки на горячей пластине. Температура горячей пластины (Sorel, модельDS37) была установлена на 55 С. У мышей наблюдали признаки дискомфорта, такие как беспокойство или перебирание лапами, попытки скрыться (спрыгнуть с пластины) или дрожание. Время реакции отмечали, когда обнаруживали один из этих признаков. Пределом для задержки ответа являлось время 15 с, чтобы предотвратить повреждения тканей лап. Для определения течения времени обезболивания мышей наблюдали в различные интервалы времени после введения соединения (или растворителя, или стандарта). Интервалы времени обычно были 30, 60 или 120 мин (или иные). Для каждого записанного времени среднее время реакции контрольной группы умножали на 1,5. Время реакции каждого обработанного животного сравнивали со "средним контролем 1,5". Если время реакции было ниже "среднего контроля 1,5", считали, что у мыши не было обезболивающего эффекта. Если время реакции было выше "среднего контроля 1,5", считали,что у мыши был обезболивающий эффект. Если процент мышей, демонстрирующих обезболивание, был ниже, чем 30%, соединение считали неактивным. Фиг. 1-3 показывают обезболивающие эффекты соединения 9 у мышей по оценке реакции мыши в тесте на горячей пластине и ингибирования ответа судорогами в PBQ-анализе. Как показано на фиг. 1, спустя 15 мин время задержки ответа у мышей, обработанных 2,5 мг/кг соединения 9, является почти макси 21 мальным. Время реакции у мышей, обработанных 5 мг/кг соединения 9, составляет около 19 с по сравнению с контрольным значением в примерно 7 с. Эти результаты показывают, что соединение 9 действительно дозозависимо увеличивает задержку ответа на источник тепла. Фиг. 2 показывает ингибирование ответа судорогами, усиленное у мышей оральным введением соединения 9 за один час до введенияPBQ. Фиг. 3 демонстрирует ингибирование ответа судорогами, усиленное у мышей подкожным введением соединения 9 за двадцать минут перед введением PBQ. В обеих фигурах дозозависимое ингибирование ответа судорогами наблюдали для соединения 9, введенного и оральным, и подкожным способом. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) где R1 является Н, С 1-6 алкилом или С 6-12 арилом,который может быть замещен группами -СООН,-NH2 или гуанидином;R2 и R3 являются, независимо друг от друга, Н, ОН, C1-6 алкилом, -C(NH)-NH2, положительно заряженной группой или С 7-13 аралкилом,который может быть замещен NH2, ОН, C1-6 алкилом или галогеном; илиR2 и R3 совместно образуют 5- или 6 членное кольцо, которое может содержать гетероатом;R5 является, независимо, Н, C1-6 алкилом или С 7-13 аралкилом, который может быть прерван одним или более гетероатомом(атомами);n является целым числом от 0 до 2;m является целым числом от 0 до 3. 2. Соединение по п.1, отличающееся тем,что Х выбран из S и SO2, m равно 3 и n равно 0. 3. Соединение по п.2, отличающееся тем,что Х является S. 4. Соединение по п.3, отличающееся тем,что R2 является Н и R3 выбран из Н, ОН и-C(NH)-NH2. 5. Соединение по п.3, отличающееся тем,что R1 является метилом. 6. Соединение по п.3, отличающееся тем,что R4 выбран из ОН и метокси. 7. Соединение по п.6, отличающееся тем,что R4 является ОН. 8. Соединение по п.1, выбранное из(-)-транс-5,6,7,8,9,11,12-гептагидро 10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина. 10. Способ обезболивания млекопитающего путем введения ему фармацевтически эффективного количества соединения формулы (I) по п.1. 11. Способ по п.10, отличающийся тем, что Х выбран из S и SO2, m равно 3 и n равно 0. 12. Способ по п.11, отличающийся тем, что Х является S. 13. Способ по п.10, отличающийся тем, чтоR1 является метилом и R4 является ОН. 15. Способ по п.10, отличающийся тем, что вышеупомянутое соединение выбрано из: 8b: 5,6,7,8,9,11,12-гептагидро-3-метокси 5-метил-10-тиа-5,11-метанобензоциклодецен 13-амина; 9: 5,6,7,8,9,11,12-гептагидро-3-гидрокси 5-метил-10-тиа-5,11-метанобензоциклодецен 13-амина (сульфазоцин); 10: 5,6,7,8,9,11,12-гептагидро-3-гидрокси 5-метил-10-тиа-5,11-метанобензоциклодецен 13-гидроксиламина; 8 а: 5,6,7,8,9,11,12-гептагидро-3-метокси 5-метил-10-тиа-5,11-метанобензоциклодецен 13-гидроксиламина; 9 а: 9b: (+)-транс-5,6,7,8,9,11,12-гептагидро 10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина; 11: транс-5,6,7,8,9,11,12-гептагидро-10 тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-гуанидина; 12: транс-5,6,7,8,9,11,12-гептагидро-10 сульфоно-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина. 16. Способ активации опиоидных рецепторов млекопитающего путем введения ему соединения формулы (I) по п.1, активирующего опиоидный рецептор. 17. Способ по п.16, отличающийся тем, что Х выбран из S и SО 2, m равно 3 и n равно 0. 18. Способ по п.16, отличающийся тем, чтоR1 является метилом и R4 является ОН. 20. Способ по п.16, отличающийся тем, что вышеупомянутое соединение выбрано из 8b: 5,6,7,8,9,11,12-гептагидро-3-метокси 5-метил-10-тиа-5,11-метанобензоциклодецен 13-амина; 9: 5,6,7,8,9,11,12-гептагидро-3-гидрокси 5-метил-10-тиа-5,11-метанобензоциклодецен 13-амина (сульфазоцин);(-)-транс-5,6,7,8,9,11,12-гептагидро 10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина; 9b: (+)-транс-5,6,7,8,9,11,12-гептагидро 10-тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина; 11: транс-5,6,7,8,9,11,12-гептагидро-10 тиа-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-гуанидина; 12: транс-5,6,7,8,9,11,12-гептагидро-10 сульфоно-3-гидрокси-5-метил-5,11-метанобензоциклодецен-13-амина. 21. Применение фармацевтически эффективного количества соединения формулы I по п.1 в качестве обезболивающего средства для млекопитающего. 22. Применение соединения формулы I по п.1 в качестве средства, активирующего опиоидный рецептор млекопитающего.
МПК / Метки
МПК: C07D 337/16, C07C 215/44, A61K 31/38, A61P 23/00
Метки: соединения, гетероциклические, применение, обезболивания
Код ссылки
<a href="https://eas.patents.su/13-1185-geterociklicheskie-soedineniya-dlya-obezbolivaniya-i-ih-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Гетероциклические соединения для обезболивания и их применение.</a>
Диароматические соединения пропинила или диенила, промежуточные соединения, фармацевтические и косметические композиции на основе указанных соединений и применение косметических композиций.

Номер патента: 1060
Опубликовано: 30.10.2000
Автор: Бернардон Жан-Мишель
МПК: A61P 17/00, C07C 63/66, A61K 31/192...
Метки: косметические, косметических, указанных, основе, соединений, композиции, фармацевтические, диенила, пропинила, соединения, композиций, промежуточные, диароматические, применение
Формула / Реферат:
1. Диароматические соединения пропинила или диенила общей формулы (I) где R1 является (I) радикалом -СН3, (II) радикалом -СН2-O-R6, (III) радикалом -О-R6, (IV) радикалом -CO-R7, где R6 и R7 имеют значения, приведенные ниже, Аr является радикалом, отвечающим следующей формуле (а): где R5 имеет значения, приведенные ниже, Х является радикалом формулы или или где R8 и R9 имеют значения, приведенные ниже, R2 и R3,...
4-аминотетрагидробензизоксазоловые или -изотиазоловые соединения, их применение и фармацевтическая композиция на их основе

Номер патента: 380
Опубликовано: 24.06.1999
Авторы: Перрегор Енс Кристьян, Скоусбо Арне, Крогсгор-Ларсен Повл, Мольтсен Ленс Сибюлле, Фальк Эрик, Фрёлунн Бенте
МПК: C07D 261/20
Метки: применение, 4-аминотетрагидробензизоксазоловые, изотиазоловые, фармацевтическая, соединения, основе, композиция
Формула / Реферат:
1. 4-Аминотетрагидробензизоксазоловые или -изотиазоловые соединения, имеющие общую формулу I: где R1 и R2, каждый, независимо выбран из группы, состоящей из А) водорода, С3-7 циклоалкила, фенила или группы где R, R8 и R9, каждый, независимо выбран из группы, состоящей из водорода, C1-4 алкила, C2-4 алкенила, С2-4 алкинила, С1-4 алкокси- C1-4 алкила, С3-7 циклоалкила, С3-7 циклоалкил-С1-4 алкила, фенила, фенил-C1-4 алкила, фенокси-C1-4...
Замещенные [2-(1-пиперазинил)этокси]метильные соединения, способ их получения и их применение

Номер патента: 831
Опубликовано: 24.04.2000
Авторы: Бодсон Ги, Люркин Франсуаз, Мотте Женевьев, Делирс Мишель, Дюшене Ги
МПК: C07D 295/08
Метки: 2-(1-пиперазинил)этокси]метильные, замещенные, соединения, применение, способ, получения
Формула / Реферат:
1. Замещенные [2-(1-пиперазинил)этокси]метильные соединения формулы где R1 обозначает -CONH2, -CN, -СООН, -СООМ или -СООR3, причем М обозначает щелочной металл и R3 обозначает С1-4алкил; и R2 обозначает атом водорода или группу -COR4 или R5, причем R4 выбран из групп -OR5 или R7, в которых R5 обозначает аллил или алкиларил, R6 обозначает линейный или разветвленный С1-4алкил, галоидалкил, алкиларил, алкилнитроарил или алкилгалоидарил,...
Способ селективной регуляции клеточного роста и клеточной дифференцировки и применение фармацевтической композиции, содержащей арил- и гетероарилхиназолиновые соединения для этой цели.

Номер патента: 840
Опубликовано: 24.04.2000
Авторы: Магир Мартин П., Спада Альфред П., Майерз Майкл Р., Персонз Пол Е.
МПК: A61K 31/535
Метки: дифференцировки, клеточного, применение, селективной, композиции, клеточной, цели, гетероарилхиназолиновые, содержащей, способ, фармацевтической, регуляции, роста, соединения, этой, арил
Формула / Реферат:
1. Способ селективной регуляции клеточного роста и клеточной дифференцировки, характеризующийся активностью рецептора типа 2 человеческого эпидермального фактора роста (HER2), причем указанный способ предусматривает введение пациенту, нуждающемуся в такой регуляции, эффективного HER2-ингибирующего количества соединения формулы где А представляет замещенную или незамещенную моно- или бициклическую арильную, гетероарильную, циклоалкильную или...
Щелочной детергент с высоким содержанием неионного поверхностно-активного вещества и комплексообразователя и применение амфотерного соединения в качестве солюбилизатора для этого детергента

Номер патента: 105
Опубликовано: 27.08.1998
Авторы: Хаммарстранд Карин, Скёльд Рольф, Карлссон Гунвор
МПК: C11D 1/72
Метки: солюбилизатора, вещества, соединения, качестве, применение, детергент, неионного, щелочной, амфотерного, детергента, поверхностно-активного, этого, высоким, комплексообразователя, содержанием
Формула / Реферат:
1. Щелочной концентрат в виде прозрачного водного раствора, который после разбавления водой пригоден для использования в качестве детергента, отличающийся тем, что он содержит, по меньшей мере, 4 мас.% неионного алкоксилатного поверхностно-активного вещества, которое содержит 2-12, предпочтительно 3-10 алкиленоксигрупп, имеющих 2-4 атома углерода, причем, по меньшей мере, 50% алкиленоксигрупп являются этиленоксигруппами, по меньшей мере, 13...
Предыдущий патент: Роторно-турбинный двигатель внутреннего сгорания ю.м.лужкова
Следующий патент: Нетоксичный, недорогой буровой раствор
Случайный патент: Регулируемое нагревающее устройство для водонагревателя