Имидазолкарбоксамиды
Номер патента: 18434
Опубликовано: 30.07.2013
Авторы: Шкерянтц Джеффри Майкл, Мэйхью Дэниэл Рей, Чжан Дэи, Хилевич Альберт, Лю Бинь











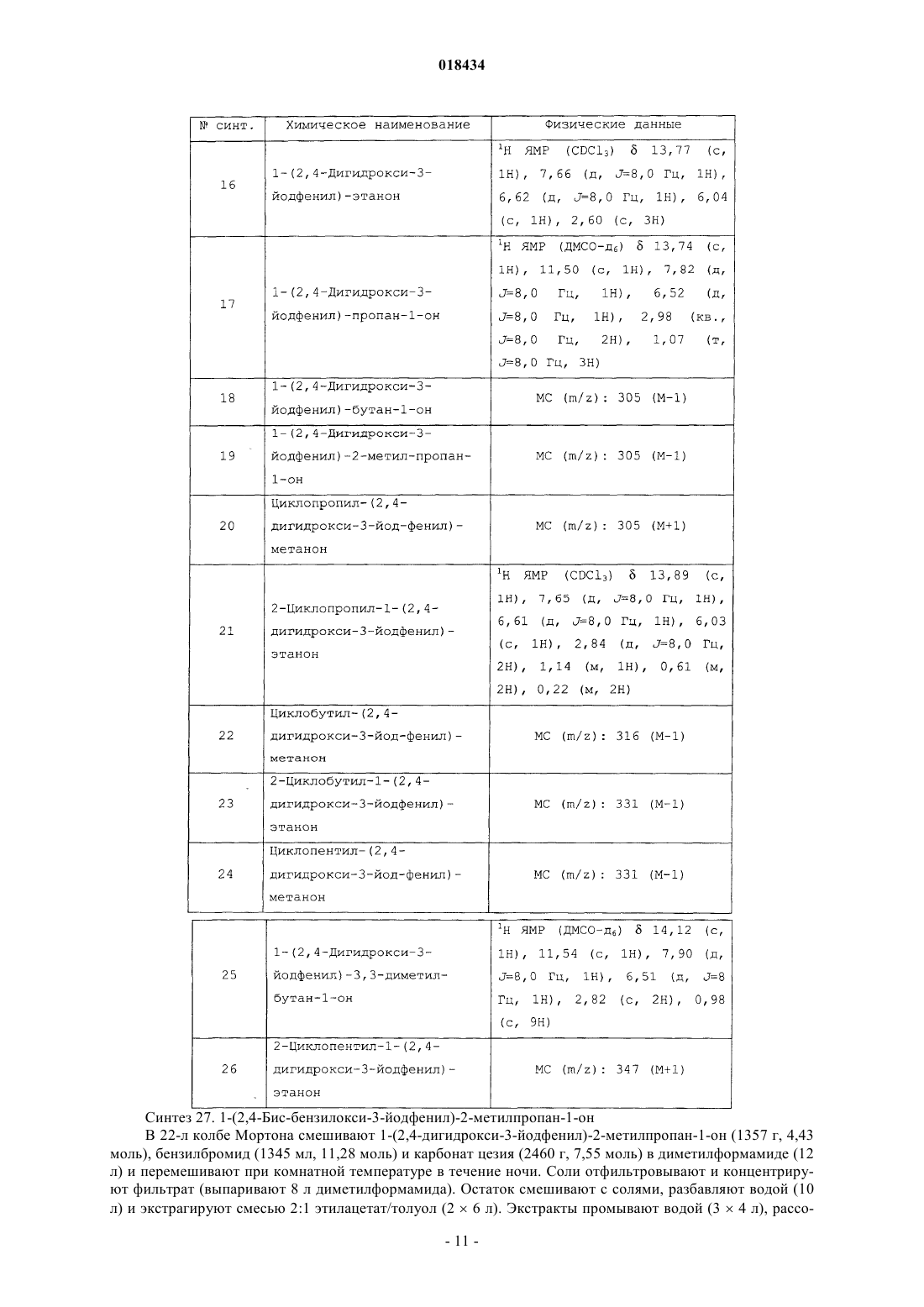


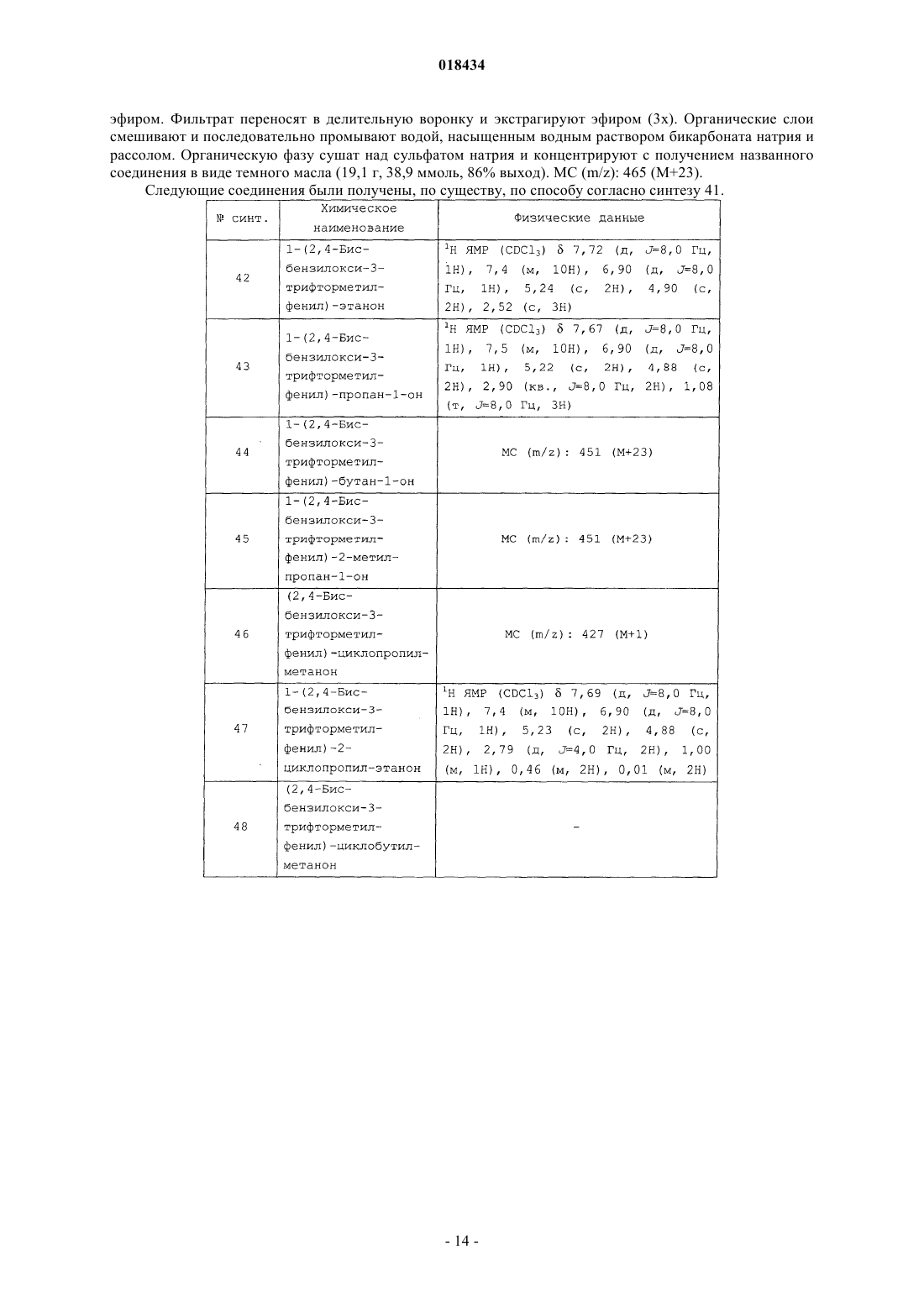












Формула / Реферат
1. Соединение формулы I или его фармацевтически приемлемая соль

где R1 представляет собой C1-C5алкил, С3-С5циклоалкил или С3-С5циклоалкилметил;
R2 представляет собой С1-С3алкил, хлор, бром, фтор или трифторметил;
А выбран из группы, состоящей из

R3 представляет собой водород или метил;
R4 представляет собой водород или С1-С3 алкил и
R5 представляет собой один заместитель, выбранный из группы, состоящей из водорода, метила, метокси, хлора и фтора, или два заместителя, каждый из которых представляет собой фтор.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где А представляет собой

3. Соединение по п.1 или 2, представляющее собой соединение формулы Ia или его фармацевтически приемлемую соль

где R1 представляет собой C1-C5алкил, С3-С5циклоалкил или С3-С5циклоалкилметил;
R2 представляет собой C1-C3алкил, бром или трифторметил;
R3 представляет собой водород или метил;
R4 представляет собой водород или С1-С3алкил и
R5 представляет собой один заместитель, выбранный из группы, состоящей из водорода, метила, метокси, хлора и фтора, или два заместителя, каждый из которых представляет собой фтор.
4. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где R1 представляет собой C1-C5алкил.
5. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где R2 представляет собой трифторметил.
6. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где R3 представляет собой водород.
7. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где R4 представляет собой C1-C3алкил.
8. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где R5 представляет собой водород, метил или метокси.
9. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где R5 представляет собой водород.
10. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где R4 представляет собой метил.
11. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где R1 представляет собой изопропил.
12. Соединение по п.1 или его фармацевтически приемлемая соль, где
R1 представляет собой метил, этил, пропил, изопропил, изобутил, 2,2-диметилпропил, циклопропил, циклобутил, циклопентил, циклопропилметил, циклобутилметил или циклопентилметил;
R2 представляет собой метил, трифторметил или бром;
А выбран из группы, состоящей из

R3 представляет собой водород или метил и
R4 представляет собой водород, метил, этил или изопропил.
13. Соединение по любому из предшествующих пунктов, представляющее собой 4-(3-гидрокси-4-изобутирил-2-трифторметилфеноксиметил)бензиламид 1-метил-1Н-имидазол-4-карбоновой кислоты или его фармацевтически приемлемую соль.
14. Соединение по любому из предшествующих пунктов, представляющее собой 4-(3-гидрокси-4-изобутирил-2-трифторметилфеноксиметил)бензиламид 1-метил-1Н-имидазол-4-карбоновой кислоты.
15. Фармацевтическая композиция, содержащая соединение по любому из пп.1-14 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель.
16. Применение соединения по любому из пп.1-14 или его фармацевтически приемлемой соли для лечения депрессии.
17. Способ лечения депрессии, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-14 или его фармацевтически приемлемой соли.
Текст
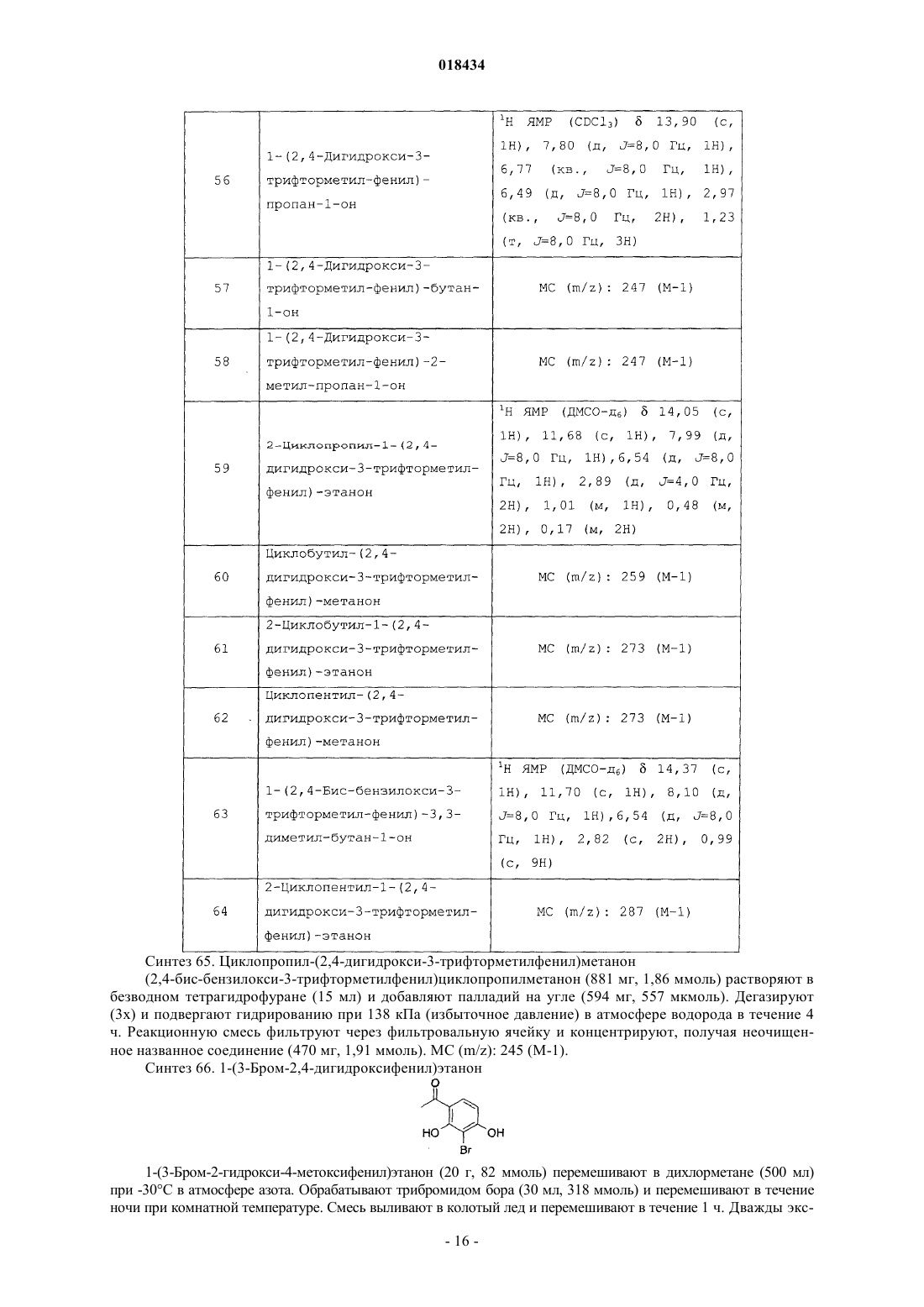
(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) В настоящем изобретении предложены некоторые производные имидазолкарбоксамидов,фармацевтические композиции на их основе, способы их применения. 018434 В настоящем изобретении предложены некоторые производные имидазолкарбоксамида, фармацевтические композиции на их основе, способы их применения и способы их получения.L-глутамат представляет собой основной возбуждающий нейромедиатор центральной нервной системы, называемый возбуждающей аминокислотой. Глутаматные рецепторы включают два основных подтипа: ионотропные рецепторы (лиганд-зависимые ионные каналы) и сопряженные с G-белком метаботропные рецепторы, содержащие семь трансмембранных доменов (mGluRs). Семейство метаботропных рецепторов включает восемь рецепторов, которые подразделяют на три группы на основании схожести их последовательностей, типа передачи сигнала и фармакологии. Рецепторы группы I (mGluR1 и mGluR5 и их сплайс-варианты) положительно связаны с гидролизом инозитфосфата и индукцией внутриклеточного кальциевого сигнала. Рецепторы группы II (mGluR2 и mGluR3) и рецепторы группы III (mGluR4,mGluR6, mGluR7 и mGluR8) отрицательно связаны с аденилатциклазой и регулируют уровни циклического аденозинмонофосфата путем непрямого подавления активности аденилатциклазы. Указанные подтипы рецептора mGlu характеризуются уникальными типами экспрессии в центральной нервной системе,которые могут быть мишенью для новых селективных средств. В публикации международной заявки на патент WO2004/057869 А 1 предложены некоторые производные ацетофенона в качестве агентов, оказывающих потенцирующее действие на рецепторыmGluR2, и антагонистов рецепторов CysLT1, а также отмечается, что эти соединения можно применять для лечения ряда состояний, включая депрессию, тревожность и мигрень. В заявке на Европейской патент ЕР 0516069 предложены некоторые производные ацетофенона в качестве антагонистов лейкотриена В 4, а также указано, что эти соединения можно применять для лечения аллергии, ревматоидного артрита и воспалительного заболевания кишечника. Соединения согласно настоящему изобретению представляют собой селективные агенты, оказывающие потенцирующее действие на метаботропные рецепторы группы II, в частности рецептор mGluR2(mGluR2) , в особенности по отношению к mGluR1, mGluR3, mGluR4, mGluR5 и mGluR8. Полагают, что указанные соединения как таковые подходят для лечения состояний, связанных с рецептором mGluR2,например, депрессии, включая большое депрессивное расстройство, тревожности, включая генерализированное тревожное расстройство, а также депрессии, сопровождаемой тревожностью (тревожнодепрессивное расстройство смешанного типа), включая большое депрессивное расстройство, сопровождаемое генерализированным тревожным расстройством. Таким образом, в настоящем изобретении предложены новые соединения, оказывающие потенцирующее действие на mGluR2, при этом, как полагают, указанные соединения как таковые можно применять для лечения вышеупомянутых нарушений. Такие новые соединения могут восполнить потребность в безопасных и эффективных лекарственных средствах для лечения состояний, связанных с указанными рецепторами, не вызывая побочных эффектов. В настоящем изобретении предложено соединение формулы I или его фармацевтически приемлемая сольR3 представляет собой водород или метил;R4 представляет собой водород или C1-C3 алкил иR5 представляет собой один заместитель, выбранный из группы, состоящей из водорода, метила,метокси, хлора и фтора; или два заместителя, каждый из которых представляет собой фтор. В настоящем изобретении также предложена фармацевтическая композиция, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель. В настоящем изобретении также предложено соединение согласно настоящему изобретению или его фармацевтически приемлемая соль для применения в терапии. В настоящем изобретении также предложено соединение согласно настоящему изобретению или его фармацевтически приемлемая соль для применения для лечения депрессии.-1 018434 В настоящем изобретении также предложено применение соединения согласно настоящему изобретению или его фармацевтически приемлемой соли для получения лекарственного средства для лечения депрессии. В настоящем изобретении также предложен способ лечения депрессии, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Конкретное соединение формулы I представляет собой соединение, в котором -А- представляет собой Конкретное соединение формулы I представляет собой соединение формулы Ia или его фармацевтически приемлемую сольR3 представляет собой водород или метил;R4 представляет собой водород или С 1-С 3 алкил иR5 представляет собой один заместитель, выбранный из группы, состоящей из водорода, метила,метокси, хлора и фтора; или два заместителя, каждый из которых представляет собой фтор. Конкретное соединение формулы I или Ia представляет собой соединение, в котором R1 представляет собой С 1-С 5 алкил. Конкретное соединение формулы I или Ia представляет собой соединение, в котором R2 представляет собой трифторметил. Конкретное соединение формулы I или Ia представляет собой соединение, в котором R3 представляет собой водород. Конкретное соединение формулы I или Ia представляет собой соединение, в котором R4 представляет собой C1-С 3 алкил. Конкретное соединение формулы I или Ia представляет собой соединение, в котором R5 представляет собой водород, метил или метокси. Конкретное соединение формулы I или Ia представляет собой соединение, в которомR5 представляет собой водород, метил или метокси. Более конкретное соединение формулы I или Ia представляет собой соединение, в котором R5 представляет собой водород. Более конкретное соединение формулы I или Ia представляет собой соединение, в котором R4 представляет собой метил. Более конкретное соединение формулы I или Ia представляет собой соединение, в котором R1 представляет собой изопропил. Более конкретное соединение формулы I или Ia представляет собой соединение, в которомR4 представляет собой метил иR5 представляет собой водород. Более конкретное соединение формулы I представляет собой соединение, в которомR3 представляет собой водород или метил иR4 представляет собой водород, метил, этил или изопропил. Предпочтительное соединение формулы I или Ia представляет собой 4-(3-гидрокси-4-изобутирил-2 трифторметилфеноксиметил)бензиламид 1-метил-1 Н-имидазол-4-карбоновой кислоты или его фармацевтически приемлемую соль. Более предпочтительное соединение формулы I или Ia представляет собой 4-(3-гидрокси-4 изобутирил-2-трифторметилфеноксиметил)бензиламид 1-метил-1 Н-имидазол-4-карбоновой кислоты. Другой вариант реализации настоящего изобретения включает способ получения соединения формулы I или его фармацевтически приемлемой соли, включающий: А) для соединения формулы I, где R4 представляет собой С 1-С 3 алкил реакцию сочетания соединения формулы II с R4-имидазолкарбоновой кислотой, в которой R4 представляет собой C1-С 3 алкил или В) для соединения формулы I, в котором R4 представляет собой Н,удаление защитной группы из соединения формулы VIII, в котором Pg представляет собой защитную группу в имидазоле, например трифенилметил; после чего, если необходимо получение фармацевтически приемлемой соли соединения формулы I,ее получают по реакции соединения формулы I, имеющего основной характер, с физиологически приемлемой кислотой или другим традиционным способом. Следует понимать, что соединения согласно настоящему изобретению могут существовать в виде стереоизомеров. В то время как настоящее изобретение включает все энантиомеры, диастереомеры и их смеси, предпочтительные варианты реализации представляют собой индивидуальные диастереомеры, а более предпочтительные варианты реализации представляют собой индивидуальные энантиомеры. Следует понимать, что соединения согласно настоящему изобретению могут существовать в виде таутомерных форм. Если существуют таутомерные формы, то каждая из форм, а также их смеси, находятся в рамках настоящего изобретения. Например, если группа R4 представляет собой водород, то соединение формулы I может существовать в виде таутомерных форм I и II. В этом случае следует пони-3 018434 мать, что любое упоминание соединения формулы I, в котором группа R4 представляет собой водород, в качестве таутомерной формы I включает таутомерную форму II, а также смеси форм I и II. Термин "фармацевтически приемлемая соль" включает соли присоединения кислот, существование которых связано с основной частью соединения формулы I. Такие соли включают фармацевтически приемлемые соли, перечисленные в руководстве HANDBOOK OF PHARMACEUTICAL SALTS: PROPERTIES, SELECTION AND USE, P. H. Stahl and С G. Wermuth (Eds.), Wiley-VCH, New York, 2002, известные специалистам в данной области техники. Помимо фармацевтически приемлемых солей изобретение включает и другие соли. Они могут выступать в качестве промежуточных соединений при очистке соединений или при получении других фармацевтически приемлемых солей, или подходить для целей идентификации, определения характеристик или очистки. Полагают, что соединения согласно изобретению подходят для применения в случаях, когда имеются показания для потенцирования рецептора mGluR2. В частности, полагают, что соединения согласно изобретению подходят для лечения депрессии, включая большое депрессивное расстройство, тревожности, включая генерализированное тревожное расстройство, а также депрессии, сопровождаемой тревожностью (смешанного тревожно-депрессивного расстройства). Соответственно, один из конкретных аспектов изобретения относится к лечению смешанного тревожно-депрессивного расстройства, включая большое депрессивное расстройство, сопровождаемое генерализированным тревожным расстройством. В настоящем описании термин "пациент" означает теплокровное животное, например млекопитающее, и включает человека. Очевидно также, что специалист в данной области техники может воздействовать на депрессивное расстройство путем лечения пациента, у которого в настоящий момент наблюдаются соответствующие симптомы, с помощью эффективного количества соединения формулы I. Таким образом, термины "лечение" и "лечить" включают все способы, которые обеспечивают замедление, временное прекращение,приостановку, контролирование или остановку прогрессирования нарушения и/или его симптомов, но не обязательно означают полное устранение всех симптомов. Очевидно также, что специалист в данной области техники может воздействовать на депрессивное расстройство путем лечения пациента, у которого имеется риск развития симптомов в будущем, с помощью эффективного количества соединения формулы I, при этом лечение также включает профилактическую терапию указанного расстройства. В настоящем описании термин "эффективное количество" соединения формулы I означает количество, то есть дозировку, которая эффективна при лечении депрессивного расстройства, рассматриваемого в настоящем описании. Лечащий врач-диагност без труда может определить эффективное количество с помощью традиционных методик и анализа результатов, полученных при аналогичных обстоятельствах. При определении эффективного количества, т.е. дозы соединения формулы I, лечащий врач-диагност должен учитывать ряд факторов, включая, но не ограничиваясь ими, вводимое соединение формулы I; совместное введение других средств, если таковые применяют; вид млекопитающего, его размер, возраст и общее состояние здоровья; степень вовлечения в патологический процесс или степень тяжести депрессии; ответную реакцию конкретного пациента; способ введения; характеристики биодоступности вводимого препарата; выбранный режим дозирования; применение других сопутствующих лекарственных средств и другие имеющие значение обстоятельства. Полагают, что эффективное количество соединения формулы I варьируется от примерно 0,01 миллиграмма на килограмм массы тела в сутки (мг/кг/сутки) до примерно 5 мг/кг/сутки. Предпочтительные количества могут быть определены специалистом в данной области техники. Соединения согласно настоящему изобретению можно вводить в отдельности или в виде фармацевтической композиции, то есть в сочетании с фармацевтически приемлемыми носителями или наполнителями, соотношение и природа которых определяются растворимостью и химическими свойствами,включая стабильность выбранного соединения, выбранным способом введения и стандартной фармацевтической практикой. Для удобства проведения кристаллизации, повышения растворимости и т.д. соединения согласно настоящему изобретению, несмотря на их эффективность при применении в отдельности,могут быть приготовлены в виде композиций и введены в виде соответствующих фармацевтически приемлемых солей. Таким образом, в настоящем изобретении предложены фармацевтические композиции, содержащие соединение формулы I и фармацевтически приемлемый носитель, разбавитель или наполнитель. Специалист в области техники приготовления фармацевтических композиций без труда может вы-4 018434 брать нужную форму и способ введения в зависимости от конкретных характеристик выбранного соединения, нарушения или состояния, подвергаемого лечению, стадии развития нарушения или состояния и других имеющих значение обстоятельств (REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY, 19-е издание, Mack Publishing Co. (1995. Функциональная активность in vitro по отношению к рецептору человека mGluR2 Для данных исследований применяли клеточные линии, стабильно экспрессирующие человеческий рецептор mGluR2 (см., например, Desai, Burnett, Mayne, Schoepp, Mol. Pharmacol. 48, 64 8-657, 19 95),трансфицированные совместно с переносчиком глутамата ЕААТ 1 (переносчик 1 возбуждающей аминокислоты (Excitatory Amino Acid Transporter 1 крысы и субъединицей G15. Экспрессия G15 обеспечивает возможность сопряжения этих Gi-сопряженных рецепторов с фосфолипазой С, что позволяет проводить измерения активации рецептора при помощи флюорометрического исследования кальциевого ответа. Клеточную линию поддерживали путем культивирования в модифицированной по Иглу среде Дульбекко (Dulbecco's Modified Eagle's Medium (DMEM с высоким содержанием глюкозы и гидрохлорида пиридоксина с добавлением 5% диализированной и дезактивированной нагреванием эмбриональной бычьей сыворотки, 1 мМ пирувата натрия, 10 мМ HEPES (4-(2-гидроксиэтил)-1 пиперазинэтансульфокислоты), 1 мМ L-глютамина и 5 мкг/мл бластицидина. Культуры, образующие непрерывный слой, пересевали раз в две недели, используя не содержащий фермента раствор для диссоциации. Клетки собирали за 24 ч до анализа и помещали в 96-луночные планшеты с черными стенками,покрытые поли-D-лизином, при плотности 85 К на лунку в среду, содержащую только 250 мкМ Lглутамина (свежедобавленного). Внутриклеточные уровни кальция измеряли до и после добавления соединений при помощи флюорометрического считывателя планшетов (Fluorometric Imaging Plate Reader (FLIPR), Molecular Devices,Union City, CA, США). Буферный раствор для анализа содержал сбалансированный солевой раствор Хенкса (Hank's Buffered Salt Solution (HBSS с добавлением 20 мМ HEPES. Среду удаляли, и клетки инкубировали с 8 мкМ реактива Fluo-3 АМ (Molecular Probes, Eugene, OR, США, F-1241; 50 мкл на лунку) в буферном растворе для анализа в течение 90 мин при 25 С. Раствор красителя удаляли с помощью постукивания по планшетам и заменяли ее свежим буферным раствором для анализа (50 мкл на лунку). Перед каждым экспериментом проводили анализ FLIPR при однократном добавлении (single-additionFLIPR), получая кривую доза-ответ, включающую 11 точек, для агониста глутамата. Результаты анализировали (GraphPad Prism v4, программное обеспечение Graphpad, LaJolla, CA, США), вычисляя концентрации глутамата, необходимые для индуцирования ответных ЕС 10. Соединения тестировали с помощью анализа FLIPR с двукратным добавлением (two-additionFLIPR) с применением профиля концентрация-ответ, включающий 10 точек, начиная с конечной концентрации, составляющей 25 мкМ (режим агониста) или 12,5 мкМ (режим потенцирующего агента). После серии трехкратных разбавлений в диметилсульфоксиде (ДМСО) выполняли однократное разбавление в буферном растворе для анализа. После первого считывания флуоресценции планшета с клетками в течение 5 с в планшет добавляли соединение (50 мкл на лунку). Для определения агонистической активности данные регистрировали каждую секунду в течение первых 30 с и затем каждые 3 с, а затем, спустя приблизительно 90 с было произведено второе добавление. Второе добавление состояло из 100 мкл глутамата в буферном растворе для анализа (обычно приблизительно 1 мкМ) и вызывало ответ, составляющий 10% от контрольных значений (ЕС 10). После второго добавления данные регистрировали каждые 3 с в течение приблизительно 90 с. Максимальный ответ определяли как ответ, вызываемый ECmax (100 мкМ глутамата). Воздействие,оказываемое соединением, измеряли как разность между максимальной и минимальной высотой пика в относительных единицах флуоресценции (О.Е.Ф.) за вычетом фона флуоресценции, измеряемого в отсутствие глутамата. Определения проводили, используя единичные планшеты. Действие агониста количественно определяли в виде процента стимулирования, вызываемого соединением, взятым в отдельности, относительно максимального ответа, вызываемого глутаматом. Потенцирующее действие количественно определяли в виде процентного повышения ЕС ответа агониста относительно ответа ECmax. Все данные вычисляли относительно значений ЕС 50 при помощи четырехпараметрической программы построения логистических кривых (ActivityBase v5.3.1,22, IBS, Alamenda, CA, США). Значения ЕС 50 соединений, приведенных в настоящем описании, полученные в ходе указанного анализа, составляли менее 150 нМ по отношению к mGluR2. Например, значение ЕС 50 соединения согласно примеру 1, измеренное по отношению к mGluR2, составляло 22,5 нМ. Это показывает, что соединения согласно настоящему изобретению представляют собой высокоактивные потенцирующие агенты по отношению к рецептору mGluR2. Положительное дифференциальное подкрепление условного сигнала у крыс Программу дифференциального подкрепления условного сигнала низкого уровня - 72 с (DRL-72) широко используют в качестве скринингового исследования клинически эффективных антидепрессантов, включая имипрамин, флуоксетин, пароксетин и другие средства (Sokolowski J.D., Seiden L.S. 1999.rate 72-second operant schedule in the rat. Psychopharm (Berl). 147(2) :153-61). Ряд положительных результатов, полученных при проведении этого анализа, указывает на то, что соединения, имеющие различные механизмы воздействия, обнаруживаемые при клинических испытаниях на людях, также являются эффективными и в системе согласно настоящему исследованию. Кроме того, также было обнаружено, что ингибиторы обратного захвата серотонина, например флуоксетин, соединения, ингибирующие захват норэпинефрина, например имипрамин или ребоксетин, также оказывают положительное воздействие в ходе данного исследования. Полагают, что соединения, демонстрирующие положительные результаты при дифференциальном подкреплении условного сигнала у животных, подходят для лечения депрессии у человека. Самцов крыс Хольтцмана-Спрага-Доули (Holtzman Sprague-Dawley) ограничивали в потреблении воды (перед проведением доступ к воде был в течение 20 мин в сутки) и тренировали надавливать рычаг для 4-секундного доступа к 0,025 мл воды при каждом правильном ответе во время ежедневных 60 минутных опытов. Все опыты проводили только по рабочим дням недели. После успешного прохождения тренировок на надавливание рычага крыс стали обучать в рамках DRL-24-секундного протокола, в соответствии с которым только те нажатия рычага, которые были разделены 24-секундным промежутком времени, получали подкрепление. После достижения стабильной ответной реакции в DRL-24-секундном протоколе крыс стали обучать в рамках DRL-72-секундного протокола до достижения стабильной ответной реакции с приблизительно 15%-ной эффективностью. В частности, крысы получали подкрепление за каждый ответ, выданный спустя по меньшей мере 72 с после предыдущего ответа (IRT, individual response time - индивидуальное время ответа). При ответах с IRT менее 72 с крысы не получали подкрепления, и требование IRT вновь устанавливали на 72 с. Эффективность ответа регистрировали как отношение количества ответов с подкреплением к общему количеству ответов. После достижения стабильной базисной линии для ответов, определяемой в виде количества ответов в трех предыдущих последовательных сериях опытов при не более чем 10% отклонении, начинали испытания лекарственных средств на животных. Лекарственные средства вводили животным не более 1 раза в неделю. Полученные данные включают количество выданных ответов, количество полученных подкреплений и IRT. Положительное действие лекарственного средства включает снижение общего количества ответов, повышение количества полученных подкреплений и упорядоченный сдвиг вправо кривой распределения IRT, что указывает на более эффективное приближение поведения животных к 72-секундному интервалу без потери стимульного контроля, подразумеваемого протоколом. Таблица А среднеквадратическая ошибка В ходе указанного анализа соединение согласно примеру 1 демонстрировало повышение количества полученных подкреплений и значительное снижение количества ответов при дозе 10 мг/кг (табл. А). Это показывает, что соединение согласно настоящему изобретению подходит для лечения депрессии в модели депрессии in vivo. Снижение обусловленной стрессом гипертермии у крыс Гипертермия, представляющая собой подъем внутренней температуры тела, представляет собой общее явление, которое, как было убедительно показано для множества млекопитающих, включая человека, развивается в ответ на стрессовое воздействие. Многие тревожные расстройства сопровождаются гипертермией, которая является частью патологического процесса и рассматривается как симптом заболевания. Полагают, что соединения, снижающие обусловленную стрессом гипертермию у животных,можно применять для лечения тревожных нарушений у человека. Традиционный и требующий минимального вмешательства способ анализа обусловленной стрессом гипертермии состоит в измерении температуры тела и ее повышения, вызванного стрессом, при помощи ректального термометра. Опыты проводили на самцах крыс Фишера F-344 (Harlan, Indianapolis, IN,USA), масса тела которых составляла 275-350 г. Всех животных помещали в индивидуальные помещения с автоматической подачей воды и свободным доступом к пище и воде и содержали при 12-часовом световом цикле (свет включали в 06:00). Животных не кормили приблизительно в течение 12-18 ч до начала эксперимента, который проводили во время дневной фазы цикла. Крысам перорально вводили дозируемый объем 2 мл/кг исследуемых соединений в диапазоне 1, 3, 10 и 30 мг/кг (суспендированных в 1% карбоксиметилцеллюлозе с добавлением 0,25% полисорбата 80 и 0,05% противовспенивающего агента). В качестве образца сравнения применяли антагонист mGluR5 МТЕР (3-[(2-метил-1,3-тиазол-4-6 018434 ил)этинил]пиридин), который проявлял устойчивую анксиолитическую активность в преклинических исследованиях (10 мг/кг, перорально, растворенный в воде). Сразу после проведения дозирования крыс возвращали в свои клетки, и экспериментатор выключал свет и выходил из комнаты. Комната, в которой происходило введение соединений, оставалась в темноте еще в течение 4-часового периода предварительной обработки. После периода предварительной обработки крыс на короткое время выносили поодиночке в ярко освещенную соседнюю комнату, в которой измеряли базовую температуру тела с помощью введения ректального зонда, смазанного минеральным маслом. Температуру тела измеряли с помощью микротермометра PHYSITEMP ВАТ-12, снабженного ректальным зондом для крыс PHYSITEMP RET-2(Physitemp Instruments Inc., Clifton, NJ, США). Зонд вводили приблизительно на 2 см в задний проход,измеряя внутреннюю температуру тела (это базовая температура тела, Т 1, градусы Цельсия). Спустя десять минут измеряли вторую температуру тела (Т 2). Разность в температурах тела (Т 2-Т 1) представляла собой обусловленную стрессом гипертермическую ответную реакцию. Доза соединения, которая вызывала 35% снижение обусловленной стрессом гипертермической ответной реакции по сравнению с реакцией при введении носителя, представляет собой дозу Т 35. В приведенном выше исследовании соединение согласно примеру 1 вызывало снижение обусловленной стрессом гипертермии при дозе Т 35=3,2 мг/кг. Это показывает, что соединения согласно настоящему изобретению подходят для применения в модели тревожности in vivo. Соединение формулы I может быть получено по способам, которые включают способы, известные в области химии, для получения структурно аналогичных соединений, или по способу, приведенному в настоящем описании, включая способы, приведенные в синтезах и примерах. Новый способ, предложенный в настоящем описании, представляет собой другой аспект изобретения. Способ получения соединения формулы I или его фармацевтически приемлемой соли и новые промежуточные соединения для получения соединения формулы I представляют собой дополнительные признаки настоящего изобретения и проиллюстрированы с помощью следующих методик, в которых, если не указано иное, значения радикалов являются такими, как определено выше. Схема А В общем случае, соединение формулы I может быть получено из соединения формулы II. Более конкретно, как показано на схеме А, амин формулы II подвергают реакции сочетания с имидазолкарбоновой кислотой, 1-гидроксибензотриазолом и карбодиимидным реагентом в подходящем растворителе,например тетрагидрофуране, получая амид формулы I, в котором R4 представляет собой C1-С 3 алкил. Подходящие карбодиимидные реагенты включают гидрохлорид 1-(3-диметиламинопропил)-3 этилкарбодиимида. Для амида формулы I, в котором R4 представляет собой водород, реакцию сочетания проводят с имидазолкарбоновой кислотой, в которой R4, представляющий собой водород, заменен на подходящую защитную группу, например, трифенилметил. Удаление защитной группы приводит к получению амида формулы I, в котором R4 представляет собой водород. Соединение формулы I также может быть получено из соединения формулы III. Более конкретно,как показано на Схеме В, фенол формулы III подвергают взаимодействию в условиях реакции Мицунобу(Mitsunobu) с соединением формулы IV, где X представляет собой ОН в присутствии органического фосфина, например, трифенилфосфина, и подходящего азодикарбонильного реагента, например, диизопропилазодикарбоксилата, получая соединение формулы I. Подходящие растворители включают толуол. В альтернативном случае соединение формулы I может быть получено по реакции соединения формулыIII с соединением формулы IV, в котором X представляет собой уходящую группу, в присутствии основания, например, карбоната лития, и в подходящем растворителе, например, ДМФА. Подходящие уходящие группы включают галогениды, например йодид или бромид, и эфиры сульфонатов, например метансульфонатный эфир. Схема С Соединение формулы II может быть получено из соединения формулы VII. Более конкретно, как показано на Схеме С, соединение формулы VII, в котором Y представляет собой цианогруппу или азидометил, вводят в реакцию с подходящим восстановителем, получая соединение формулы VII, в которомY представляет собой аминометил. Защита аминометильной группы приводит к образованию соединения формулы VI, которое затем вводят в реакцию с восстановителем, например, алюмогидридом лития, в растворителе, например, тетрагидрофуране, с получением гидроксильного соединения формулы V. Далее, гидроксильное соединение формулы V может быть непосредственно введено в реакцию с соединением формулы III или превращено в подходящую уходящую группу перед проведением реакции с соединением III с образованием соединения формулы II, как показано на схеме В. В следующих иллюстративных синтезах и примерах используют следующие значения и аббревиатуры: ДМСО, диметилсульфоксид (полностью дейтерированный [-Д 6] для ЯМР); МС, масс-спектр;EtOAc, этилацетат; ТГФ, тетрагидрофуран; мин, минуты; ЖХВР, жидкостная хроматография высокого давления; ЖХ-МС, ЖХВР-масс-спектрография; ГХ, газовая хроматография; МеОН, метанол; МТВЕ, метил-трет-бутиловый эфир; SCX-2, катионообменная смола; т.пл., температура плавления; и ЯМР, ядерная магниторезонансная спектроскопия или спектр. Реагенты были получены из ряда коммерческих источников. Растворители в общем случае удаляли при пониженном давлении (испаряли). В некоторых синтезах указанные выходы представляют собой представительные значения выхода для неочищенных продуктов, которые выделяли испарением или фильтрованием и использовали непосредственно без дополнительной очистки. ЖХВР проводили на приборе Waters 600 с использованием программного обеспечения Empower,версия 2; колонка: Chromolith Performance RP-18e, 4,6100 мм; расход: 5 мл/мин; градиент: 10% растворителя А (0,1% трифторуксусной кислоты в ацетонитриле), 90% растворителя В (0,1% трифторуксусной кислоты в воде) в течение 1 мин, линейное повышение до 80% растворителя А в течение 4 мин, выдержка при 80% растворителя А в течение 3 мин; детектор: Waters PDA 996 на фотодиодной матрице PDAsingle 254 channel. Синтез 1. 1-(2,4-Дигидроксифенил)-2-метилпропан-1-он Смешивают резорцин (1010 г, 9,17 моль) с трифторидэтератом бора (1,9 л, 15,0 моль) и перемешивают механическим способом в 12-л колбе Мортона при комнатной температуре. Смесь обрабатывают изобутирилхлоридом (880 мл, 8,37 моль), чистым в течение 3 ч путем добавления через капельную воронку, затем перемешивают в течение ночи при комнатной температуре. Охлажденное масло выливают в 10 кг колотого льда и экстрагируют дважды этиловым эфиром (всего 5 л). Органические слои промывают водой, насыщенным раствором хлорида натрия, сушат сульфатом магния, фильтруют, и фильтрат-8 018434 выпаривают, получая названное соединение (1636 г, количественный выход) в виде красноватого масла. ЖХВР Rt=4,29 мин; 1 Н ЯМР (CDCl3)13,03 (с, 1 Н), 7,69 (д, J = 12,0 Гц, 1 Н), 6,4 (м, 2 Н), 3,51 (гепт, J = 8,0 Гц, 1 Н), 1,23(д, J = 8,0 Гц, 6 Н). Следующее соединение было получено по существу по способу согласно синтезу 1. Изовалериановую кислоту (5,10 г, 50 ммоль, 5,5 мл) добавляют при комнатной температуре в атмосфере аргона одной порцией к смеси резорцина (5,00 г, 45,4 ммоль) и трифторидэтерата бора (38,67 г,272,5 ммоль, 34,5 мл). Реакционную смесь нагревают при 90 С в течение 1,5 ч. Реакционную смесь охлаждают до комнатной температуры, выливают в 20% водный раствор ацетата натрия и перемешивают в течение ночи. Водную смесь экстрагируют этилацетатом (3 х). Органические слои смешивают и последовательно промывают насыщенным водным раствором бикарбоната натрия и рассолом. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют под уменьшенным давлением, получая продукт в виде коричневатого масла (9,80 г; 50,5 ммоль, 111% выход). МС (m/z): 195 (М+1). Следующие соединения были получены, по существу, по способу согласно Синтезу 3. Синтез 11. 1-Бром-2,4-бис-(трет-бутилдиметилсиланилокси)бензол 4-Бромрезорцин перемешивают (50 г, 265 ммоль) в толуоле (1 л) и обрабатывают третбутилдиметилхлорсиланом (90 г, 597 ммоль) и 1 Н-имидазолом (50 г, 734 ммоль). Нагревают до кипения с обратным холодильником в течение 6 ч, затем перемешивают в течение ночи при комнатной температуре. Органический слой промывают водой, разбавленным раствором гидроксида натрия и рассолом,сушат сульфатом магния, фильтруют и выпаривают, получая названное соединение в виде янтарного масла (88,3 г, 80% выход). 1 Н ЯМР (CDCl3)7,31 (д, J = 8,0 Гц, 1 Н), 6,37 (м, 2 Н), 1,04 (с, 6 Н), 0,97 (с, 6 Н), 0,24 (с, 3 Н), 0,19 (с,3 Н). Циклопропануксусную кислоту (30 г, 300 ммоль) перемешивают в дихлорметане (1 л) и медленно обрабатывают 1,1'-карбонилдиимидазолом (52,5 г, 324 ммоль). Перемешивают в течение 2 ч при комнатной температуре и обрабатывают гидрохлоридом N,О-диметилгидроксиламина (30 г, 308 ммоль), чистым, одной порцией; перемешивают в течение ночи. Смесь выливают в воду и дважды экстрагируют дихлорметаном. Органические слои промывают водой, разбавленной соляной кислотой, насыщенным раствором бикарбоната натрия, сушат сульфатом магния, фильтруют и выпаривают, получая названное соединение (38 г, 89%). 1H ЯМР (CDCl3)3,66 (с, 3 Н), 3,18 (с, 3 Н), 2,35 (д, J=8,0 Гц, 2 Н), 1,08 (м, 1 Н), 0,54 (м, 2 Н), 0,16 (м,2 Н). Синтез 13. 2-Циклопропил-1-(2,4-дигидроксифенил)этанон 1-Бром-2,4-бис-(трет-бутилдиметилсиланилокси)бензол (100 г, 240 ммоль) перемешивают в диэтиловом эфире (1 л, безводный) при -60 С и обрабатывают 1,7 М трет-бутиллитием в пентане (290 мл, 493 ммоль), прикапывая через капельную воронку в течение 10 мин. Перемешивают в течение 15 мин, затем обрабатывают 2-циклопропил-N-метокси-N-метилацетамидом (34 г, 237 ммоль) в минимальном количестве этилового эфира. Охлаждающую баню убирают, перемешивают в течение 1 ч, обрабатывают 1 Н водной соляной кислотой (100 мл) и перемешивают еще один час. Слои разделяют, и органический слой промывают 1 Н водной соляной кислотой (100 мл), рассолом, сушат сульфатом магния, фильтруют и выпаривают. Остаток растворяют в тетрагидрофуране (1 л), обрабатывают 1 Н раствором фторида тетрабутиламмония в тетрагидрофуране (500 мл, 500 ммоль), и перемешивают в течение 3 ч. Смесь выливают в 1 л воды, содержащей 120 мл 5 Н соляной кислоты, слои разделяют, и водный слой дважды экстрагируют этилацетатом. Органические слои промывают рассолом, сушат сульфатом магния, фильтруют и выпаривают. Остаток хроматографируют на силикагеле (BiotageRadial Compression, снабженный 75-л колонкой), элюируя смесью 25% этилацетата в гексанах, получая названное соединение (43 г, 94% выход). 1(6 л) перемешивают механическим способом в двух отдельных 22-л колбах Мортона. Каждую суспензию обрабатывают йодатом калия (170 г, 794 ммоль) и йодом (410 г, 1,62 моль) и оставляют перемешиваться в течение ночи при комнатной температуре. Смеси разбавляют водой (2 л), доводят рН до 4 добавлением 5 Н соляной кислоты, затем дважды экстрагируют 4 л этилацетата. Органические слои промывают один раз разбавленным водным бисульфитом натрия, рассолом, сушат сульфатом магния, фильтруют, смешивают оба фильтрата и выпаривают до получения 2,6 кг черного твердого вещества. Хроматографируют порциями твердое вещество по 500 г на 3,5 кг силикагеля, элюируя смесью 80% дихлорметана в гептане,получая названное соединение (1357 г, 49% выход) с приблизительно 97% чистотой (данные ЖХВР: 97% чистота, 3% 3,5-дийодного аналога; данные ЖХМС это подтверждают. ЖХВР Rt=4,75 мин; 1(80 мл) добавляют при комнатной температуре йод (4,99 г,19,7 ммоль) и йодат калия (2,16 г, 10,1 ммоль). Реакционную смесь энергично перемешивают в течение ночи. Реакционную смесь разбавляют водой и экстрагируют смесь этилацетатом (3 х). Органические слои смешивают, промывают рассолом, сушат над сульфатом натрия, фильтруют и концентрируют под уменьшенным давлением, получая продукт в виде масла (15,00 г, 46,9 ммоль, 93% выход). МС (m/z): 321 (М+1). Следующие соединения были получены по существу по способу согласно синтезу 15. Синтез 27. 1-(2,4-Бис-бензилокси-3-йодфенил)-2-метилпропан-1-он В 22-л колбе Мортона смешивают 1-(2,4-дигидрокси-3-йодфенил)-2-метилпропан-1-он (1357 г, 4,43 моль), бензилбромид (1345 мл, 11,28 моль) и карбонат цезия (2460 г, 7,55 моль) в диметилформамиде (12 л) и перемешивают при комнатной температуре в течение ночи. Соли отфильтровывают и концентрируют фильтрат (выпаривают 8 л диметилформамида). Остаток смешивают с солями, разбавляют водой (10 л) и экстрагируют смесью 2:1 этилацетат/толуол (26 л). Экстракты промывают водой (34 л), рассо- 11018434 лом, сушат сульфатом магния, фильтруют и выпаривают. Полученное твердое вещество перемешивают в 9 л 10% этилацетата в гексане в течение 1 ч, фильтруют, твердое вещество промывают гексанами и сушат на воздухе, получая названное соединение (1710 г, 79% выход) в виде беловатого твердого вещества. ЖХВР Rt=6,60 мин; 1 К раствору 1- (2,4-дигидрокси-3-йодфенил)-3-метилбутан-1-она (15,00 г, 46,9 ммоль) в безводном диметилформамиде (120 мл) при комнатной температуре в атмосфере аргона добавляют бензилбромид(17,63 г, 103,1 ммоль, 12,3 мл) и карбонат цезия (30,53 г, 93,7 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Смесь фильтруют, и фильтрат концентрируют под уменьшенным давлением с получением масла. Масло разделяют между эфиром и водой. Органический слой отделяют, и водный слой экстрагируют эфиром (2 х). Органические слои смешивают, промывают рассолом, сушат над сульфатом натрия, фильтруют и концентрируют, получая красноватое масло (22,5 г, 45 ммоль, 96% выход). МС (m/z): 501 (М+1). Следующие соединения были получены, по существу, по способу согласно синтезу 28. Синтез 40. 1-(2,4-Бис-бензилокси-3-трифторметилфенил)-2-метил-пропан-1-он В 22-л колбе Мортона перемешивают 1-(2,4-бис-бензилокси-3-йодфенил)-2-метилпропан-1-он(1622 г, 8,44 моль), триамидом гексаметилфосфорной кислоты (1475 мл, 8,47 моль) и йодидом меди(I)(890 г, 4,67 моль). Дегазируют азотом через трубку для ввода, снабженную стеклянным фильтром, в течение 1 ч, затем нагревают до 80 С в течение 17 ч и перемешивают в течение выходных при комнатной температуре. Раствор отфильтровывают от соли и фильтрат концентрируют под уменьшенным давлением (удаляют 10 л растворителя). Соли промывают этилацетатом (4 л), промывные воды смешивают с концентрированным остатком и смесь переносят в 22-л делительную воронку. Смесь далее разбавляют толуолом (2 л) и промывают водой (8 л), содержащей гидроксид аммония (500 мл, конц.) и хлорид аммония (250 г). Слои разделяют, и водный слой экстрагируют смесью 2:1 этилацетат/толуол (6 л). Органические слои смешивают и промывают водой (4 л), содержащей гидроксид аммония (50 0 мл, конц.) и хлорид аммония (250 г), водой (4 л), насыщенным водным хлоридом натрия (4 л), сушат сульфатом магния,фильтруют, и фильтрат выпаривают, получая названное соединение (1460 г, 99% выход) в виде красноватого масла. ЖХВР Rt=6,51 мин; 1 Н ЯМР (ДМСО-д 6)7,79 (д, J=8,0 Гц, 1 Н), 7,4 (м, 11 Н), 5,31 (с, 2 Н), 4,81 (с, 2 Н), 3,38 (гепт, J=8,0 Гц, 1 Н), 1,00 (д, J=8,0 Гц, 6 Н); 19F ЯМР (ДМСО-д 6)-54,40 В альтернативном варианте соединение синтеза 40 может быть получено при замене триамида гексаметилфосфорной кислоты диметилформамидом. Синтез 41. 1-(2,4-Бис-бензилокси-3-трифторметилфенил)-3-метилбутан-1-он К раствору 1- (2,4-бис-бензилокси-3-йодфенил)-3-метилбутан-1-она (22,5 г, 45 ммоль) в диметилацетамиде (158 мл) и триамиде гексаметилфосфорной кислоты (22 мл) добавляют метилдифтор(фторсульфонил)ацетат (23,32 г, 121,4 ммоль, 15,4 мл) и йодид меди(I) (12,85 г, 67,5 ммоль). Через реакционную смесь в течение 5 мин пропускают аргон. Реакционную смесь нагревают при 80 С в течение ночи. Охлаждают до комнатной температуры, твердые вещества отфильтровывают и промывают- 13018434 эфиром. Фильтрат переносят в делительную воронку и экстрагируют эфиром (3 х). Органические слои смешивают и последовательно промывают водой, насыщенным водным раствором бикарбоната натрия и рассолом. Органическую фазу сушат над сульфатом натрия и концентрируют с получением названного соединения в виде темного масла (19,1 г, 38,9 ммоль, 86% выход). МС (m/z): 465 (М+23). Следующие соединения были получены, по существу, по способу согласно синтезу 41. Синтез 53. 1-(2,4-Дигидрокси-3-трифторметилфенил)-2-метилпропан-1-он 1-(2,4-бис-бензилокси-3-трифторметилфенил)-2-метилпропан-1-он (1460 г, 3,07 моль) перемешивают в диметилсульфиде (8 л, 108,81 моль), обрабатывают метансульфоновой кислотой (2,5 л, 38,13 моль),затем нагревают с обратным холодильником при слабом кипении в течение ночи. Смесь охлаждают до 33 С и обрабатывают колотым льдом (6 кг) с такой скоростью, чтобы смесь не кипела. Смесь переносят в делительную воронку, слои разделяют и водный слой экстрагируют этилацетатом (6 л). Органические слои промывают водой (4 л), насыщенным раствором хлорида натрия (4 л), сушат сульфатом магния,фильтруют и выпаривают, получая коричневое твердое вещество. Твердое вещество перекристаллизовывают из толуола (4 л), получая 527 г 1-(2,4-дигидрокси-3-трифторметилфенил)-2-метилпропан-1-она в виде желтоватого твердого вещества (3 порции). Фильтрат концентрируют до получения 380 г черного с вкраплениями твердых веществ масла и фильтруют через слой силикагеля, получая 216 г желтого твердого вещества, которое перекристаллизовывают из толуола, дополнительно получая еще 69 г материала. Все порции смешивают, получая названное соединение (596 г, 78% выход): ЖХВР Rt=4,98 мин; 1 Н ЯМР (ДМСО-д 6)14,15 (с, 1 Н), 11,70 (с, 1 Н), 8,06 (д, J=8,0 Гц, 1 Н), 6,57 (д, J=8,0 Гц, 1 Н), 3,62 К раствору 1-(2,4-бис-бензилокси-3-трифторметилфенил)-3-метилбутан-1-она (19,1 г, 38,9 ммоль) в диметилсульфиде (146 мл) при комнатной температуре в атмосфере аргона добавляют метансульфоновую кислоту (41 мл). Реакционную смесь осторожно нагревают до кипения с обратным холодильником в течение ночи. Реакционную смесь гасят водой со льдом и перемешивают в течение 1 ч. Органический слой отделяют и водный слой экстрагируют этилацетатом (3 х). Органические слои смешивают, промывают рассолом, сушат над сульфатом натрия и концентрируют, получая неочищенное масло. Полученное неочищенное масло очищают колоночной хроматографией (силикагель), элюируя смесью 25% этилацетат/гексан, получая названное соединение в виде коричневатого твердого вещества (5,3 г, 20,2 ммоль,52% выход). МС (m/z): 263 (М+1). Следующие соединения были получены по существу по способу согласно синтезу 54.(2,4-бис-бензилокси-3-трифторметилфенил)циклопропилметанон (881 мг, 1,86 ммоль) растворяют в безводном тетрагидрофуране (15 мл) и добавляют палладий на угле (594 мг, 557 мкмоль). Дегазируют(3 х) и подвергают гидрированию при 138 кПа (избыточное давление) в атмосфере водорода в течение 4 ч. Реакционную смесь фильтруют через фильтровальную ячейку и концентрируют, получая неочищенное названное соединение (470 мг, 1,91 ммоль). МС (m/z): 245 (М-1). Синтез 66. 1-(3-Бром-2,4-дигидроксифенил)этанон 1-(3-Бром-2-гидрокси-4-метоксифенил)этанон (20 г, 82 ммоль) перемешивают в дихлорметане (500 мл) при -30 С в атмосфере азота. Обрабатывают трибромидом бора (30 мл, 318 ммоль) и перемешивают в течение ночи при комнатной температуре. Смесь выливают в колотый лед и перемешивают в течение 1 ч. Дважды экс- 16018434 трагируют дихлорметаном, органические слои промывают водой, насыщенным водным раствором хлорида натрия, сушат сульфатом магния, фильтруют, и фильтрат выпаривают. Остаток хроматографируют на силикагеле, элюируя смесью 2:1 дихлорметан/гексан, получая названное соединение (13,8 г, 74% выход) в виде беловатого порошка. 1 Н ЯМР (CDCl3)13,5 (с, 1 Н), 7,65 (д, J = 8,0 Гц, 1 Н), 6,62 (д, J = 8,0 Гц, 1 Н), 6,20 (с, 1 Н), 2,60 (с,3 Н). Следующие соединения были получены по существу по способу согласно синтезу 66. 1,0 М раствор алюмогидрида лития в тетрагидрофуране (8 л) перемешивают в 4-горлой 22-л круглодонной колбе, нагревают до 40 С и обрабатывают 4-цианобензойной кислотой (350 г, 2,36 моль), чистой,порциями по 25 г, с такой скоростью, чтобы температура была ниже 60 С. Смесь перемешивают в течение ночи при комнатной температуре, охлаждают до 5 С на ледяной бане, обрабатывают водой (304 мл), 15% водным раствором гидроксида натрия (304 мл) и водой (912 мл) с такой скоростью, чтобы температура во время всего добавления была ниже 25 С. Ледяную баню убирают и перемешивают в течение 1 ч, затем фильтруют, осадок на фильтре промывают тетрагидрофураном (2 л). Фильтрат сушат сульфатом магния, фильтруют и фильтрат обрабатывают ди-трет-бутилдикарбонатом (576 г, 2,59 моль). Перемешивают в течение ночи при комнатной температуре, затем разбавленным этиловым эфиром (7,5 л),промывают рассолом (22 л), сушат сульфатом магния, фильтруют и выпаривают фильтрат до получения беловатого твердого вещества. Твердое вещество суспендируют в этилацетате (1 л) и гексанах (8 л) и фильтруют, получая названное соединение (457 г, 82% выход) в виде беловатого твердого вещества. 1 Н ЯМР (CDCl3)7,30 (ABq, J = 12,0, 8,0 Гц, 4 Н), 4,8 (шир.с, 1 Н), 4,68 (шир.с, 2 Н), 4,31 (д, J = 4 Гц,2 Н), 1,7 (шир.с, 1 Н), 1,46 (с, 9 Н). Синтез 71. 2-Хлор-4-метилбензойная кислота, метиловый эфир 4 Н хлороводород в 1,4-диоксане (20 мл, 80 ммоль) добавляют к 2-хлор-4-метилбензойной кислоте(5,0 г, 29,3 ммоль) в метаноле (60 мл). Реакционную смесь перемешивают при комнатной температуре в течение выходных. Реакционную смесь концентрируют и остаток разделяют между этилацетатом и насыщенным водным раствором бикарбоната натрия. Органический слой отделяют, и водный слой экстрагируют этилацетатом (2 х). Органические слои смешивают, промывают рассолом, сушат над сульфатом натрия и концентрируют, получая названное соединение в виде коричневатого масла (4,7 г, 25,5 ммоль). Следующие соединения были получены по существу по способу согласно синтезу 71. Бензоилпероксид (308 мг, 1,27 ммоль) добавляют к смеси метил-3-хлор-4-метилбензоата (4,7 г, 25,5 ммоль) и N-бромсукцинимида (4,98 г, 28,00 ммоль) в безводном четыреххлористом углероде (100 мл). Реакционную смесь кипятят в течение 4 ч с обратным холодильником и охлаждают до комнатной температуры. Реакционную смесь фильтруют, и фильтрат концентрируют, получая названное соединение в виде неочищенного масла (7,6 г, 28,8 ммоль). МС (m/z): 264 (М+1).- 17018434 Следующие соединения были получены по существу по способу согласно синтезу 74. Азид натрия (2,44 г, 37,4 9 ммоль) добавляют при комнатной температуре в атмосфере аргона к неочищенному метиловому эфиру 4-бромметил-2-хлорбензойной кислоты (7,6 г, 28,84 ммоль) в безводном диметилформамиде (100 мл). Реакционную смесь перемешивают при 50 С в течение ночи. Реакционную смесь гасят водой и экстрагируют эфиром (3 х). Органические слои смешивают, промывают рассолом,сушат над сульфатом натрия и концентрируют, получая неочищенный продукт (5,8 г, 25,7 ммоль). Следующие соединения были получены, по существу, по способу согласно Синтезу 78. Трифенилфосфин (30,3 г, 115 ммоль) добавляют к неочищенному метиловому эфиру 4-азидометил 2-хлорбензойной кислоты (5,8 г, 25,7 ммоль) в тетрагидрофуране (152 мл) и воде (4,6 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют, и остаток разделяют между 1 Н соляной кислотой и этилацетатом. Органический слой отделяют, и органический слой промывают водой. Водные слои смешивают,промывают этилацетатом и затем нейтрализуют 1 Н гидроксидом натрия. Полученную водную смесь экстрагируют этилацетатом (3 х). Органические слои смешивают, промывают рассолом, сушат над сульфатом натрия и концентрируют, получая неочищенное названное соединение (2,2 г, 11,0 ммоль). МС (m/z): 200 (М+1). Следующие соединения были получены, по существу, по способу согласно синтезу 83. Смесь метилового эфира 4-азидометил-3-метоксибензойной кислоты (4,1 г, 18,5 ммоль) и палладия на угле (4,9 г, 4,63 ммоль) в этаноле (200 мл) подвергают гидрированию в атмосфере водорода при 345 КПа в течение 24 ч при комнатной температуре. Реакционную смесь фильтруют через диатомитовую землю и фильтрат концентрируют, получая неочищенное названное соединение (2,9 г, 14,9 ммоль). МС(m/z): 196 (М+1). Неочищенный продукт используют на следующем этапе без дополнительной очистки. Следующее соединение было получено по прописи синтеза 86. Синтез 88. (4-Аминометил-3-фторфенил)метанол В безводный тетрагидрофуран (100 мл) в атмосфере аргона добавляют 1 М алюмогидрид лития в тетрагидрофуране (121 мл, 121 ммоль). Реакционную смесь нагревают до 40 С и в течение 1 ч добавляют небольшими порциями 4-циано-3-фторбензойную кислоту (5,0 г, 30,28 ммоль). Реакционную смесь перемешивают при 40 С в течение 4 ч и затем при комнатной температуре в течение ночи. Реакционную смесь охлаждают до 0 С и гасят, последовательно добавляя воду (5 мл), 15% раствор гидроксида натрия(17 мл) и воду (5 мл). Смесь оставляют нагреваться до комнатной температуры и перемешивают в течение 1 ч. Осадок фильтруют через диатомитовую землю и фильтрат концентрируют, получая названное соединение (4,4 г, 28,4 ммоль). Синтез 89. 4-Аминометил-3-метилбензойная кислота, метиловый эфир В автоклав Парра (Parr) загружают ацетат палладия (1,5 г, 6,5 ммоль), 1,1'бис(дифенилфосфино)ферроцен (4,4 г, 7,9 ммоль), 4-бром-2-метилбензонитрил (12,5 г, 64 ммоль), метанол (159 мл), ацетонитрил (240 мл) и триэтиламин (46 мл). Автоклав герметизируют, продувают N2 (6 х),продувают моноксидом углерода (6 х) и нагнетают давление моноксидом углерода (862 кПа). Реакционную смесь нагревают при 100 С в течение 24 ч. Реакционную смесь оставляют охлаждаться до комнатной температуры и фильтруют. Фильтрат концентрируют. Остаток (41 г) повторно растворяют в метаноле (1 л) . Добавляют 7 н раствор аммиака в метаноле (428 мл) и никель Ренея (8 мл). Продувают газообразным азотом (3 х), продувают водородом (3 х) и нагнетают давление водородом до 419 кПа. Смесь нагревают при 40 С в течение 18 ч. Реакционную смесь оставляют охлаждаться до комнатной температуры и фильтруют. Фильтрат концентрируют, получая неочищенное названное соединение (16,8 г). Синтез 90. 4-Циано-2-фторбензойная кислота, метиловый эфир К 4-циано-2-фторбензойной кислоте (10,0 г, 60,5 ммоль) в метаноле (100 мл) добавляют 4 Н хлороводород в 1,4-диоксане (100 мл, 400 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют. Остаток очищают колоночной хроматографией(силикагель), элюируя смесью 20% этилацетат/гексан, получая названное соединение (6,8 г, 38,0 ммоль). Синтез 91. 4-Аминометил-2-фторбензойная кислота, метиловый эфир К метиловому эфиру 4-циано-2-фторбензойной кислоты (5,8 г, 30,0 ммоль) в 2 Н растворе аммиака в метаноле (580 мл) добавляют суспензию никеля Ренея в этаноле (1,2 г, 1,2 мл). Продувают газообразным азотом (3 х), продувают водородом (3 х) и нагнетают давление водородом до 419 кПа. Реакционную смесь нагревают при 40 С в течение 18 ч. Реакционную смесь оставляют охлаждаться до комнатной температуры и фильтруют. Фильтрат концентрируют с получением неочищенного названного соединения (6,9 г,99%). МС (m/z): 184 (М+1). Синтез 92. 4-(трет-бутоксикарбониламинометил)-2-хлорбензойная кислота, метиловый эфир(100 мл) добавляют ди-трет-бутилдикарбонат (2,65 г, 12,1 ммоль). Реакционную смесь перемешивают при 50 С в течение 4 ч. Реакционную смесь концентрируют и разделяют между водой и этилацетатом. Органический слой отделяют, и водный слой экстрагируют этилацетатом (2 х). Органические слои смешивают, промывают рассолом, сушат над сульфатом натрия и концентрируют, получая названное соединение (3,2 г, 10,7 ммоль). МС (m/z): 322 (М+23). Следующие соединения были получены, по существу, по способу согласно синтезу 92. Синтез 100. 4-[(трет-бутоксикарбонилметиламино)метил]-3-метилбензойная кислота, метиловый эфир 60% дисперсию гидрида натрия в масле (472 мг, 11,81 ммоль) добавляют порциями при 0 С в атмосфере аргона к метиловому эфиру 4-(трет-бутоксикарбониламинометил)-3-метилбензойной кислоты (3,0 г, 10,74 ммоль) в безводном диметилформамиде (50 мл). По окончании добавления реакционную смесь перемешивают в течение 15 мин при 0 С. К реакционной смеси добавляют метилйодид (735 мкл, 11,8 ммоль). Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают в течение ночи. Реакционную смесь гасят насыщенным водным хлоридом аммония и экстрагируют этилацетатом (3 х) . Органические слои смешивают, промывают рассолом, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографией (силикагель), элюируя смесью 25% этилацетат/гексан, получая названное соединение. МС (m/z): 316 (М+23). Синтез 101. (3-Хлор-4-гидроксиметилбензил)карбаминовая кислота, трет-бутиловый эфир К безводному тетрагидрофурану (240 мл) при комнатной температуре в атмосфере аргона добавляют 1 М алюмогидрид лития в тетрагидрофуране (32 мл, 32 ммоль), охлаждают до 0 С и медленно добавляют раствор метилового эфира 4-(трет-бутоксикарбониламинометил)-2-хлорбензойной кислоты (3,2 г,10,7 ммоль) в тетрагидрофуране (120 мл). Реакционную смесь перемешивают при 0 С в течение 45 мин. Реакционную смесь гасят насыщенным водным раствором хлорида аммония и экстрагируют этилацетатом (3 х). Органические слои смешивают, промывают рассолом, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографией (силикагель), элюируя смесью 35% этилацетат/гексан, получая названное соединение (2,1 г, 7,7 ммоль). МС (m/z): 294 (М+23). Следующие соединения были получены, по существу, по способу согласно синтезу 101. Синтез 109. (3-Йодметилбензил)карбаминовая кислота, трет-бутиловый эфир В дихлорметане (45 мл) перемешивают трифенилфосфин, фиксированный на полистироле (4,27 г,12,8 ммоль), и имидазол (0,86 г, 12,7 ммоль). Добавляют раствор трет-бутилового эфира (3 гидроксиметилбензил)карбаминовой кислоты (1,70 г, 6,1 ммоль) в дихлорметане (45 мл). Тремя порциями добавляют йод (3,22 г, 12,7 ммоль). Перемешивают 16 ч. Фильтруют через слой диатомитовой земли. Промывают водным раствором тиосульфата натрия. Органическую часть сушат над сульфатом натрия. Фильтруют и концентрируют. Остаток хроматографируют на силикагеле, элюируя смесью 25% этилацетат/дихлорметан, получая названное соединение (2,53 г, 7,3 ммоль). Синтез 110. 3-(трет-бутоксикарбониламинометил)бензойная кислота, метиловый эфир В смеси дихлорметана (200 мл) и насыщенного водного бикарбоната натрия (100 мл) перемешивают гидрохлорид метилового эфира 3-аминометилбензойной кислоты (2,13 г, 10,6 ммоль). Добавляют дитрет-бутилдикарбонат (2,76 г, 12,7 ммоль) и перемешивают в течение 2 ч. Органическую фазу отделяют и сушат над сульфатом натрия. Фильтруют и концентрируют. Остаток очищают хроматографией (силикагель), элюируя смесью 20-30% этилацетат/гексан, получая названное соединение (2,8 г, 100%). Синтез 111. 3-[(трет-бутоксикарбонилметиламино)метил]бензойная кислота, метиловый эфир Метиловый эфир 3-(трет-бутоксикарбониламинометил)бензойной кислоты (2,80 г, 10,6 ммоль) растворяют в диметилформамиде (60 мл). Добавляют 60% дисперсию гидрида натрия в масле (0,52 г, 13 ммоль). Перемешивают в течение 1 ч. Добавляют метилйодид (0,81 мл, 13 ммоль) и перемешивают еще 1 ч. Реакционную смесь гасят водой и концентрируют. Остаток разделяют между этилацетатом и водой. Органический слой отделяют и сушат над сульфатом натрия. Фильтруют и концентрируют. Остаток очищают хроматографией (силикагель), элюируя смесью 15-25% этилацетат/гексан, получая названное соединение (1,70 г, 57%). Синтез 112. (3-Гидроксиметилбензил)метилкарбаминовая кислота, трет-бутиловый эфир Метиловый эфир 3-[(трет-бутоксикарбонилметиламино)метил]бензойной кислоты (1,70 г, 6,1 ммоль) растворяют в тетрагидрофуране (60 мл) при 0 С. Добавляют по каплям 1 М раствор алюмогидрида лития в тетрагидрофуране (8 мл, 8 ммоль) и перемешивают в течение 2 ч. Реакционную смесь гасят водой (3 мл), 5 Н раствором гидроксида натрия (3 мл) и сноваводой (9 мл), фильтруют и концентрируют. Остаток очищают хроматографией (силикагель), элюируя смесью 40-60% этилацетат/гексаны, получая- 21018434 названное соединение (1,42 г, 93%). Синтез 113. 6-Гидроксиметилникотиновая кислота, метиловый эфир В реакционную колбу загружают диметиловый эфир 2,5-пиридиндикарбоновой кислоты (15 г, 76,8 ммоль), хлорид кальция (34,12 г, 307,4 ммоль), этанол (100 мл) и тетрагидрофуран (100 мл). Смесь охлаждают до 0 С. К реакционной смеси медленно добавляют боргидрид натрия (3,49 г, 92,3 ммоль). Температуру реакционной смеси поддерживают равной 0 С и смесь перемешивают в течение 7 ч. Твердые вещества удаляют фильтрованием. Смесь выливают на лед. Экстрагируют дихлорметаном (100 мл 4). Органические слои смешивают и сушат сульфатом натрия. Растворитель удаляют под уменьшенным давлением, получая 9,6 г (75% выход) неочищенного продукта, метилового эфира 6-гидроксиметилникотиновой кислоты. МС (m/z): 168,3 (М+1). Синтез 114. 6-Хлорметилникотиновая кислота, метиловый эфир В колбу загружают метиловый эфир 6-гидроксиметилникотиновой кислоты (9,6 г, 57,4 ммоль) и дихлорметан (200 мл) и охлаждают до 0 С. Добавляют тионилхлорид (10,25 г, 86,14 ммоль). Смесь перемешивают при комнатной температуре в течение 1 ч. Реакционную смесь концентрируют под уменьшенным давлением, получая неочищенный метиловый эфир 6-хлорметилникотиновой кислоты (13 г,102% выход) в виде желтого твердого вещества. МС (m/z): 223 (М+1). Синтез 115. 6-Азидометилникотиновая кислота, метиловый эфир В колбу загружают метиловый эфир 6-хлорметилникотиновой кислоты (9,1 г, 40,98 ммоль) и диметилсульфоксид (100 мл) и охлаждают до 0 С. К реакционной смеси добавляют азид натрия (4 г, 61,47 ммоль) и карбонат натрия (13 г, 122,9 ммоль). Смесь оставляют медленно нагреваться до комнатной температуры. Перемешивают в течение 1 ч при комнатной температуре. К смеси добавляют воду (100 мл). Экстрагируют диэтиловым эфиром (100 мл х 3). Органический слой промывают водой и рассолом. Сушат сульфатом натрия. Растворитель удаляют под уменьшенным давлением, получая продукт (6,8 г, 86% выход) в виде светло-желтого масла. МС (m/z): 193,3 (М+1). Синтез 116. 6-Аминометилпиридин-3-илметанол В колбу загружают метиловый эфир 6-азидометилникотиновой кислоты (2,3 г, 11,97 ммоль) и тетрагидрофуран (100 мл) и охлаждают до 0 С. Медленно добавляют 1 М раствор алюмогидрида лития в тетрагидрофуране (17,95 мл, 17,95 ммоль). Смесь перемешивают в течение 30 мин. Реакционную смесь гасят льдом. Добавляют 10 мл насыщенного раствора виннокислого калия-натрия. Перемешивают в течение 30 мин. Фильтруют, растворитель удаляют под уменьшенным давлением, получая 6 аминометилпиридин-3-илметанол (1,6 г, 97% выход) в виде желтого масла. Синтез 117. (5-Гидроксиметилпиридин-2-илметил)карбаминовая кислота, трет-бутиловый эфир В колбу загружают 6-аминометилпиридин-3-илметанол (1,6 г, 11,58 ммоль) и тетрагидрофуран (50 мл). Затем к реакционной смеси добавляют ди-трет-бутилдикарбонат (3,79 г, 17,3 ммоль). Перемешивают в течение 30 мин. Концентрируют, получая остаток. Очищают продукт хроматографией (силикагель),элюируя смесью метанола в дихлорметане (от 95:5 до 9:1), получая трет-бутиловый эфир (5 гидроксиметилпиридин-2-илметил)карбаминовой кислоты (2,03 г, 73%). МС (m/z): 239,3 (М+1). Синтез 118. (3-Хлор-4-метилбензил)карбаминовая кислота, трет-бутиловый эфир К 3-хлор-4-метилбензиламину (9,9 г, 63,6 ммоль) в трет-бутиловом спирте (250 мл) при комнатной температуре добавляют ди-трет-бутилдикарбонат (15,3 г, 70,0 ммоль). Перемешивают при комнатной температуре в течение 2 ч и нагревают при 50 С в течение 3 ч. Реакционную смесь концентрируют и разделяют между водой и этилацетатом. Органический слой отделяют, и водный слой экстрагируют этилацетатом (2 х) . Органические слои смешивают, промывают рассолом, сушат над сульфатом натрия и концентрируют, получая названное соединение (16,2 г, 63,3 ммоль). МС (m/z): 278 (М+23). Синтез 119. (3-Хлор-4-метилбензил)дикарбаминовая кислота, ди-трет-бутиловый эфир К трет-бутиловому эфиру (3-хлор-4-метилбензил)карбаминовой кислоты (8,0 г, 31,3 ммоль) в безводном дихлорметане (150 мл) при комнатной температуре добавляют ди-трет-бутилдикарбонат (7,51 г,34,4 ммоль), диизопропилэтиламин (6,0 мл, 34,41 ммоль) и диметиламинопиридин (382 мг, 3,13 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют и остаток разделяют между этилацетатом и водой. Органический слой отделяют и водный слой экстрагируют этилацетатом (2 х). Органические слои смешивают, промывают рассолом, сушат над сульфатом натрия и концентрируют. Остаток очищают хроматографией (силикагель), элюируя смесью 15% этилацетат/гексан, получая названное соединение (3,3 г, 9,27 ммоль). МС (m/z): 378 (М+23). Синтез 120. (4-Бромметил-3-хлорбензил)дикарбаминовая кислота, ди-трет-бутиловый эфир К смеси ди-трет-бутилового эфира (3-хлор-4-метилбензил)дикарбаминовой кислоты (1,0 г, 2,8 ммоль) и N-бромсукцинимида (550 мг, 3,1 ммоль) в безводном четыреххлористом углероде (22 мл) при- 22018434 комнатной температуре в атмосфере аргона добавляют бензоилпероксид (34 мг, 140 ммоль). Реакционную смесь кипятят в течение 4 ч и охлаждают до комнатной температуры в течение ночи. Реакционную смесь фильтруют, и фильтрат концентрируют. Остаток очищают хроматографией (силикагель), элюируя смесью 20% этилацетат/гексан, получая названное соединение (614 мг). МС (m/z): 457 (М+23). Синтез 121. 1-Тритил-1 Н-имидазол-4-карбоновая кислота, метиловый эфир К смеси метил-4-имидазолкарбоксилата (1,0 г, 7,93 ммоль) и трифенилметилхлорида (2,43 г, 8,72 ммоль) в безводном ацетонитриле (25 мл) в течение 10 мин при комнатной температуре добавляют триэтиламин (1,88 мл, 13,48 ммоль) и перемешивают в течение ночи. Гасят водой и экстрагируют этилацетатом (3 х). Органические слои смешивают, последовательно промывают 1 Н раствором соляной кислоты,водой, насыщенным водным раствором бикарбоната натрия и рассолом, сушат над сульфатом натрия и концентрируют, получая названное соединение, метиловый эфир 1-тритил-1 Н-имидазол-4-карбоновой кислоты (2,8 г, 7,60 ммоль). Синтез 122. 1-Тритил-1 Н-имидазол-4-карбоновая кислота 1 Н гидроксид натрия (22,8 мл, 22,8 ммоль) добавляют при комнатной температуре к метиловому эфиру 1-тритил-1 Н-имидазол-4-карбоновой кислоты (2,8 г, 7,60 ммоль) в тетрагидрофуране (20 мл) и метаноле (20 мл). Реакционную смесь перемешивают при комнатной температуре в течение 5 ч. Реакционную смесь подкисляют 5 Н водным раствором соляной кислоты приблизительно до рН 6. Твердое вещество отфильтровывают и сушат, получая названное соединение в виде белого твердого вещества (2 г,5,64 ммоль). Синтез 123. 1-Изопропил-1 Н-имидазол-4-карбоновая кислота, метиловый эфир К охлажденному (0 С) раствору метилового эфира 1 Н-имидазол-4-карбоновой кислоты (6,03 г, 47,8 ммоль) в диметилформамиде (150 мл) порциями в течение 10 мин добавляют гидрид натрия (2,87 г, 71,7 ммоль, 60% в минеральном масле). Охлаждающую баню убирают и перемешивают при комнатной температуре в течение 4,5 ч. Смесь охлаждают до 0 С и по каплям в течение 10 мин добавляют изопропилйодид (8,94 г, 52,6 ммоль). Охлаждающую баню убирают и перемешивают при комнатной температуре в течение 20 ч. Смесь гасят насыщенным водным раствором хлорида аммония и экстрагируют этилацетатом (3 х). Объединенные экстракты промывают рассолом, сушат над сульфатом натрия, фильтруют и концентрируют под уменьшенным давлением, получая масло. Масло очищают колоночной хроматографией (силикагель), элюируя смесью 25% ацетон/гексаны, получая продукт в виде масла (1,41 г, 8,4 ммоль, 17% выход). М/С (m/z): 169 (М+Н). Синтез 124. 1-Изопропил-1 Н-имидазол-4-карбоновая кислота К раствору метилового эфира 1-изопропил-1 Н-имидазол-4-карбоновой кислоты (1,41 г, 8,38 ммоль) в метаноле (10 мл) и тетрагидрофуране (8 мл) добавляют раствор моногидрата гидроксида лития (1,06 г,25,1 ммоль) в воде (8 мл). Перемешивают при комнатной температуре в течение 20 ч. Концентрируют,получая твердое вещество. Твердое вещество растворяют в небольшом количестве воды и доводят 5 Н соляной кислотой до рН 4. Водный слой промывают этилацетатом и затем водный слой концентрируют,получая неочищенный продукт в виде твердого вещества (2,09 г). МС (m/z): 155 (М+Н). Синтез 125. 4-[3-Гидрокси-4-(3-метилбутирил)-2-трифторметилфеноксиметил]бензилкарбаминовая кислота, трет-бутиловый эфир 1- (2,4-дигидрокси-3-трифторметилфенил)-3-метилбутан-1-он (2 г, 7,63 ммоль), трет-бутиловый эфир (4-гидроксиметилбензил)карбаминовой кислоты (1,99 г, 8,39 ммоль) и трифенилфосфин (2,20 г,8,39 ммоль) смешивают в безводном толуоле (125 мл) при комнатной температуре в атмосфере аргона. Медленно, в течение 30 мин, добавляют диизопропил азодикарбоксилат (1,70 г, 8,39 ммоль, 1,7 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Концентрируют смесь под уменьшенным давлением и остаток очищают колоночной хроматографией (силикагель), элюируя смесью 25% этилацетат/гексан, получая продукт в виде белого твердого вещества (1,5 г, 3,12 ммоль, 41% выход). МС (m/z): 480 (М-1). Следующие соединения были получены по существу по способу согласно синтезу 125. Неочищенный ди-трет-бутиловый эфир (4-бромметил-3-хлорбензил)дикарбаминовой кислоты (614 мг) добавляют к раствору 1-(2,4-дигидрокси-3-трифторметилфенил)-2-метилпропан-1-она (385 мг, 1,55 ммоль) в безводном диметилформамиде (25 мл). К реакционной смеси добавляют карбонат лития (219 мг, 2,97 ммоль) и нагревают при 60 С в течение 20 ч. Реакционную смесь охлаждают до комнатной температуры и фильтруют. Фильтрат гасят водой и экстрагируют этилацетатом (3 х). Органические слои смешивают, промывают рассолом, сушат над сульфатом натрия и концентрируют. Остаток очищают колоночной хроматографией (силикагель), элюируя смесью 20% этилацетат/гексан, получая названное соединение (320 мг), которое все еще содержит примеси. МС (m/z): 500 (М-С 4 Н 9 ОСО). Следующее соединение было получено, по существу, по способу согласно синтезу 150.- 26018434 Растворы 1-(2,4-дигидрокси-3-трифторметилфенил)-2-метилпропан-1-она (211 г, 850,1 ммоль),трет-бутилового эфира (4-гидроксиметилбензил)карбаминовой кислоты (195 г, 821,8 ммоль) и трифенилфосфина (212 г, 808,3 ммоль) в толуоле (9 л) перемешивают механическим способом в двух отдельных 22-л колбах Мортона. Растворы обрабатывают диизопропилазодикарбоксилатом (183 мл, 923 ммоль) в течение 60 мин, затем перемешивают в течение выходных при комнатной температуре. Растворы смешивают и выпаривают до получения коричневого масла. Масло растворяют в 1,4-диоксане (8 л), обрабатывают 4 Н раствором хлороводорода в 1,4-диоксане (3 л, 12 моль) и нагревают до 92 С в течение 6 ч (во время нагревания при 52 С наблюдали выделение газа и образование осадка). Охлаждают до комнатной температуры, полученное твердое вещество отфильтровывают и промывают диоксаном, 30% диоксаном в гексанах и гексанами, получая названное соединение (510 г, 78% выход) в виде коричневатого твердого вещества. ЖХВР Rt=4,71 мин; 1 Н ЯМР (ДМСО-д 6)14,00 (с, 1 Н), 8,4 (шир, с, 3 Н), 7,50 (ABq, J = 12,0, 8,0 Гц, 4 Н), 6,92 (д, J = 8,0 Гц, 1 Н), 5,37 (с, 2 Н), 4,0 (м, 2 Н), 3,73 (гепт, J = 4,0 Гц, 1 Н), 1,12 (д, J = 4,0 Гц, 6 Н); 19 К смеси трет-бутилового эфира 4-[3-гидрокси-4-(3-метилбутирил)-2-трифторметилфеноксиметил]бензилкарбаминовой кислоты (1,4 г, 2,91 ммоль) в безводном 1,4-диоксане (25 мл) добавляют 4 М хлороводород в 1,4-диоксане (25 мл) и реакционную смесь нагревают при 50 С в течение 1,5 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом и фильтруют,получая продукт в виде белого твердого вещества (934 мг, 2,24 ммоль, 77% выход). Следующие соединения были получены, по существу, по способу согласно синтезу 153.
МПК / Метки
МПК: A61P 25/00, A61K 31/4164, C07D 233/92
Метки: имидазолкарбоксамиды
Код ссылки
<a href="https://eas.patents.su/30-18434-imidazolkarboksamidy.html" rel="bookmark" title="База патентов Евразийского Союза">Имидазолкарбоксамиды</a>
Предыдущий патент: Способ переработки парафинового масла
Следующий патент: Способ изготовления гетероструктур (варианты) для среднего ик-диапазона, гетероструктура (варианты) и светодиод и фотодиод на основе этой гетероструктуры
Случайный патент: Применение латексной композиции, содержащей по меньшей мере одну уреидогруппу, для адгезии на древесине