Термофильный микроорганизм, модифицированный для повышенной выработки этанола, и способ получения этанола при его использовании
Номер патента: 17548
Опубликовано: 30.01.2013
Авторы: Мартин Стив, Мерсье Клэр, Милнер Пол, Рудд Брайан, Аткинсон Энтони, Криппс Роджер, Элей Кирстин














Формула / Реферат
1. Термофильный микроорганизм, модифицированный для повышенной выработки этанола, в котором первая модификация представляет собой инактивацию гена лактатдегидрогеназы, а вторая модификация активирует ген пируватдегидрогеназы.
2. Микроорганизм согласно п.1, в котором вторая модификация представляет собой вставку промотора гена левее гена пируватдегидрогеназы, причем промотор работает в анаэробных условиях.
3. Микроорганизм согласно п.1 или 2, дополнительно модифицированный с целью инактивации гена дигидролипоамид-трансацетилазы.
4. Микроорганизм согласно любому из предыдущих пунктов, дополнительно модифицированный с целью инактивации гена пируватформиатлиазы.
5. Микроорганизм согласно любому из предыдущих пунктов, не содержащий системы рестрикции.
6. Микроорганизм согласно любому из предыдущих пунктов, который является спорообразующим.
7. Микроорганизм согласно любому из предыдущих пунктов, который относится к виду Geobacillus.
8. Микроорганизм по п.7, который представляет собой Geobacillus thermoglucosidasius.
9. Микроорганизм согласно любому из предыдущих пунктов, который стабилен в культуральной среде с содержанием этанола вплоть до 30% (вес./об.).
10. Микроорганизм согласно любому из предыдущих пунктов, который способен метаболизировать целлобиозу, ксилобиозу и/или крахмал, а также их олигомеры.
11. Микроорганизм согласно любому из предыдущих пунктов, который способен к росту при температуре 40-85°С, предпочтительно 50-70°С.
12. Микроорганизм согласно любому из предыдущих пунктов, который содержит ненативный ген пируватдекарбоксилазы.
13. Микроорганизм согласно любому из предыдущих пунктов, который содержит ненативный ген алкогольдегидрогеназы.
14. Микроорганизм согласно любому из предыдущих пунктов, в котором в гене лактатдегидрогеназы не содержится элемента интеграции.
15. Микроорганизм согласно любому из предыдущих пунктов, в котором нативный ген лактатдегидрогеназы или его часть были удалены.
16. Способ получения этанола, включающий культивирование микроорганизма согласно любому из предыдущих пунктов в подходящих условиях в присутствии С3, С5 или С6 сахаров или состоящих из них олигомеров.
17. Способ согласно п.16, который осуществляют при температуре в пределах 40-70°С.
18. Способ согласно п.17, который осуществляют при температуре в пределах от 52 до 65°С.
19. Способ согласно любому из пп.16-18, согласно которому рН культуральной среды находится в пределах от 4 до 7,5.
20. Источник корма для животных, включающий микроорганизм согласно любому из пп.1-15.
Текст
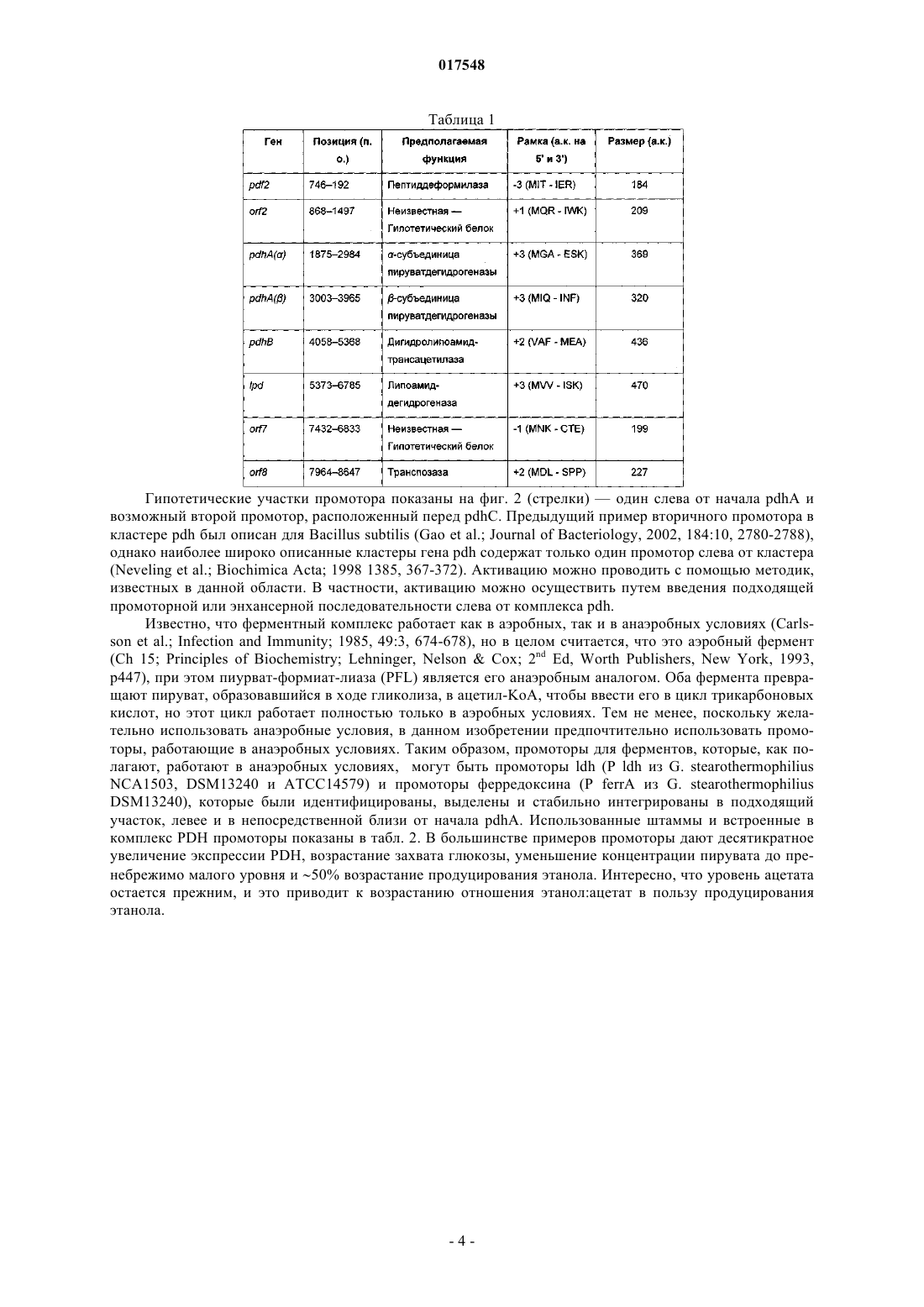
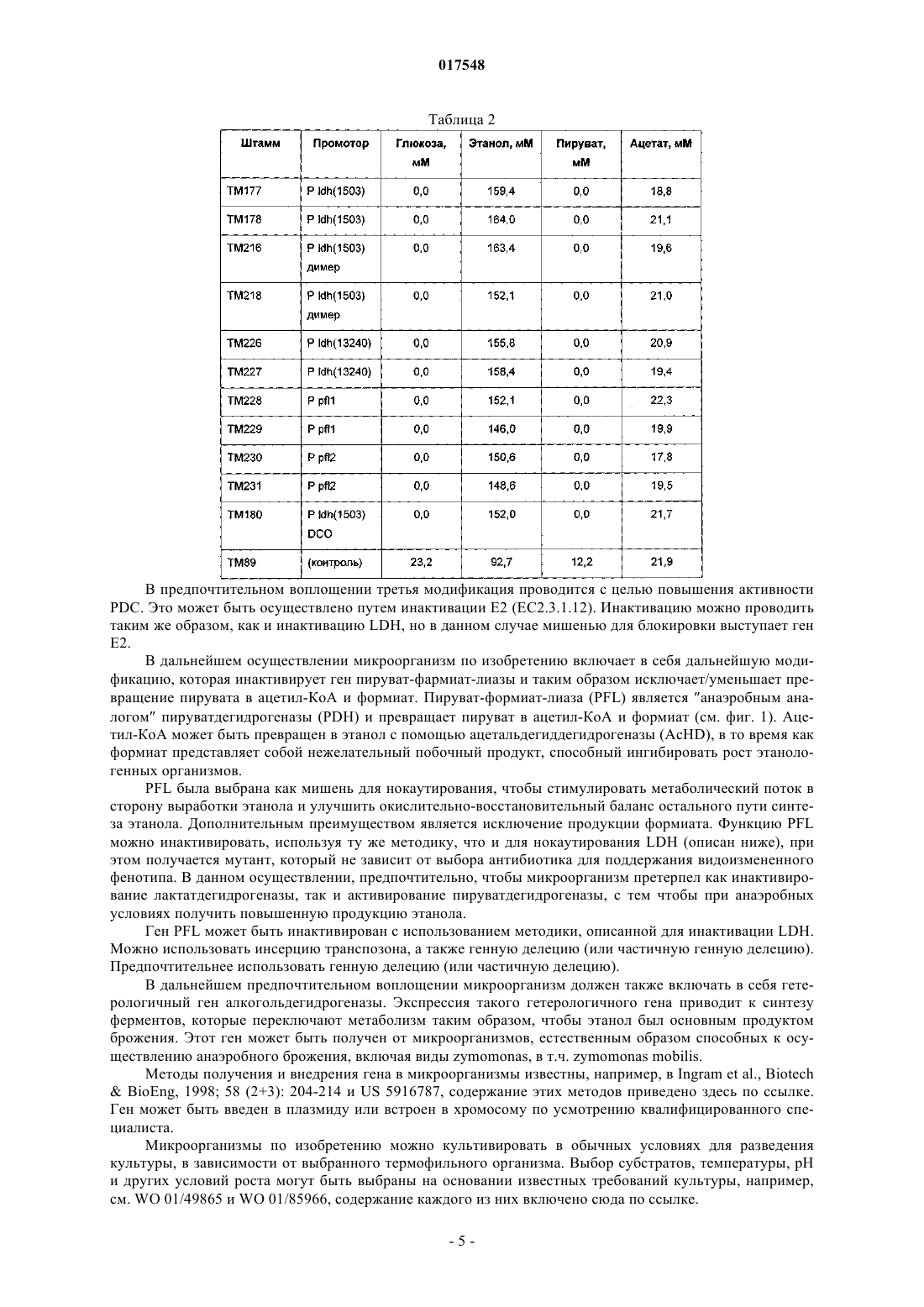
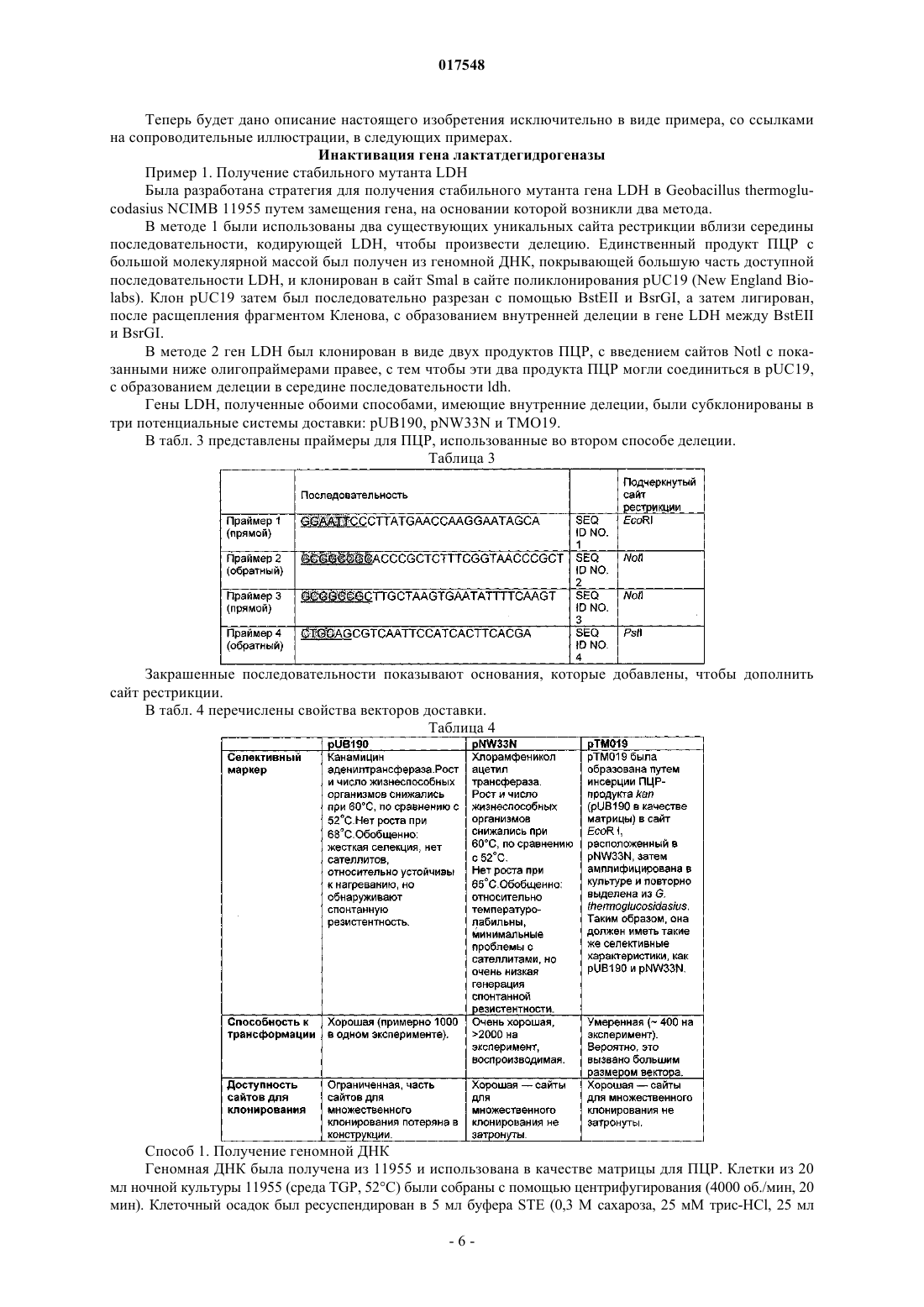
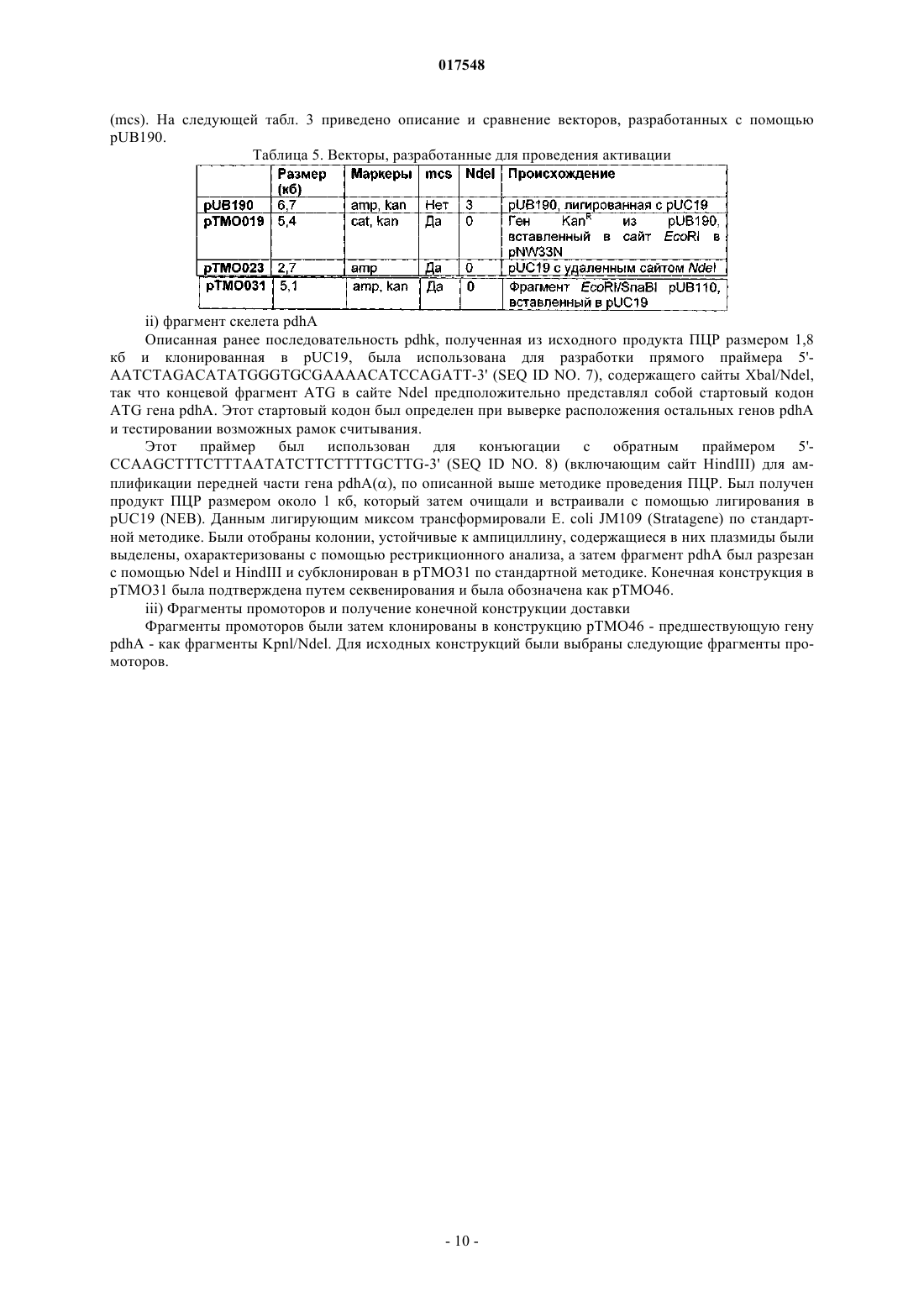
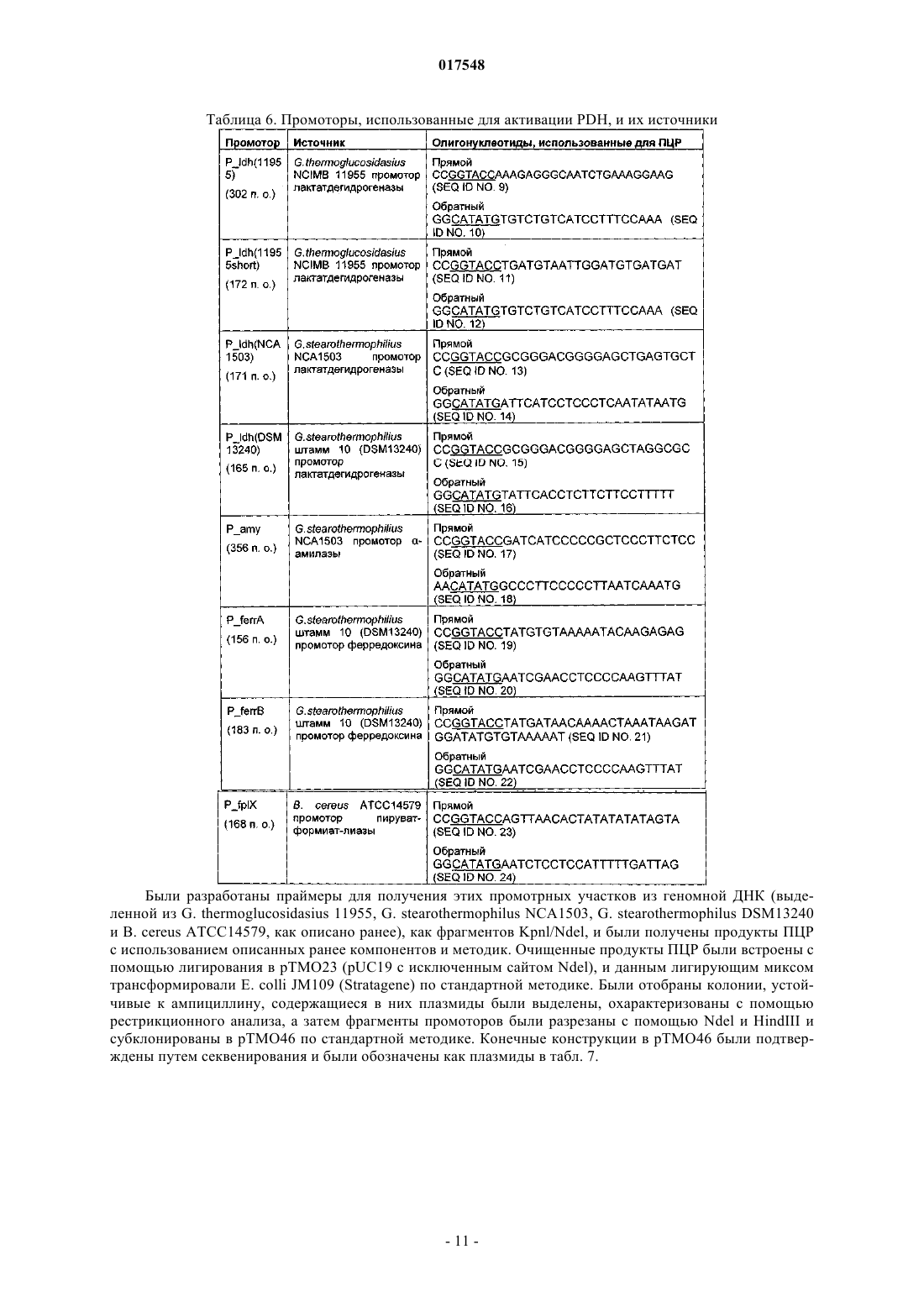
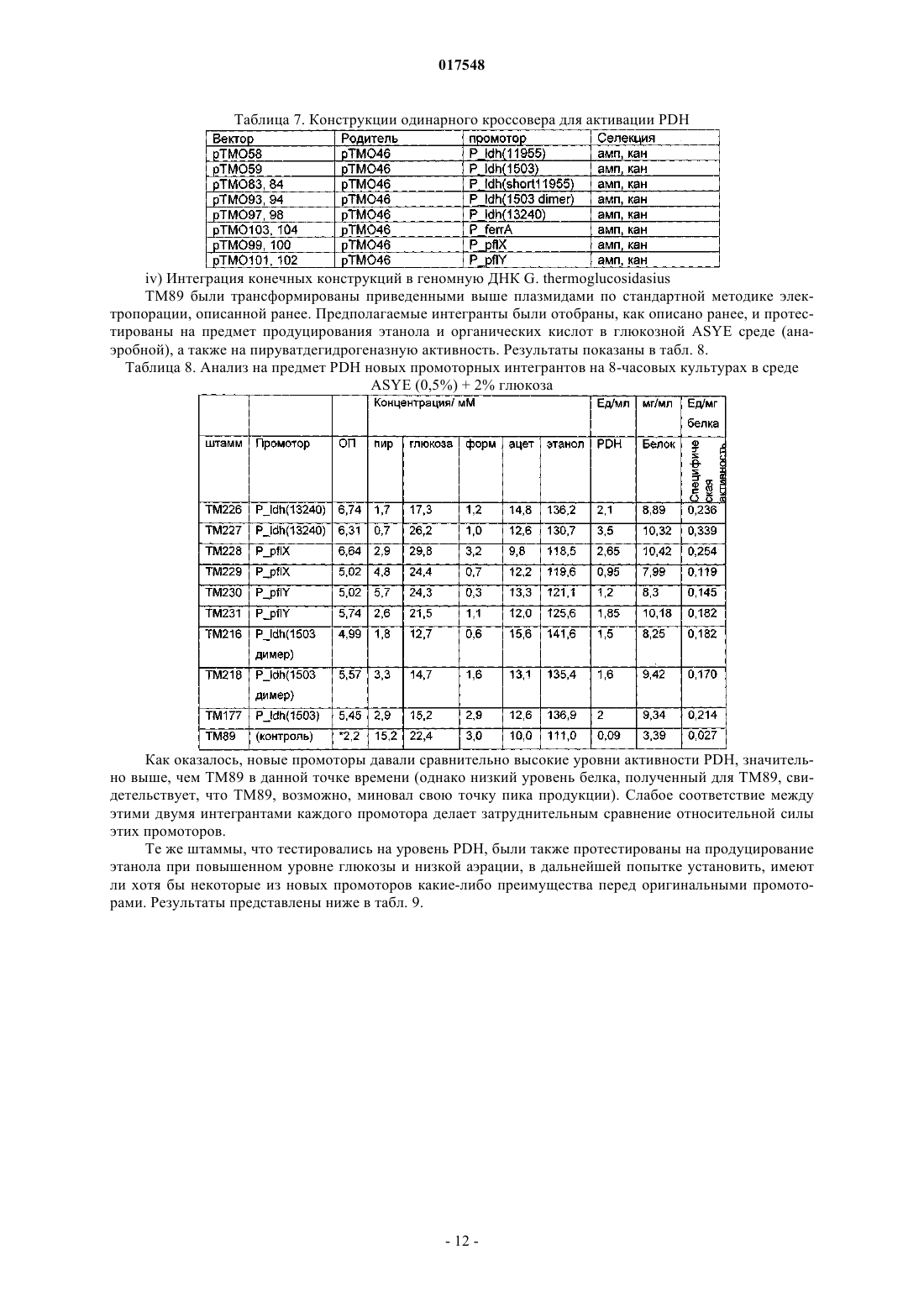
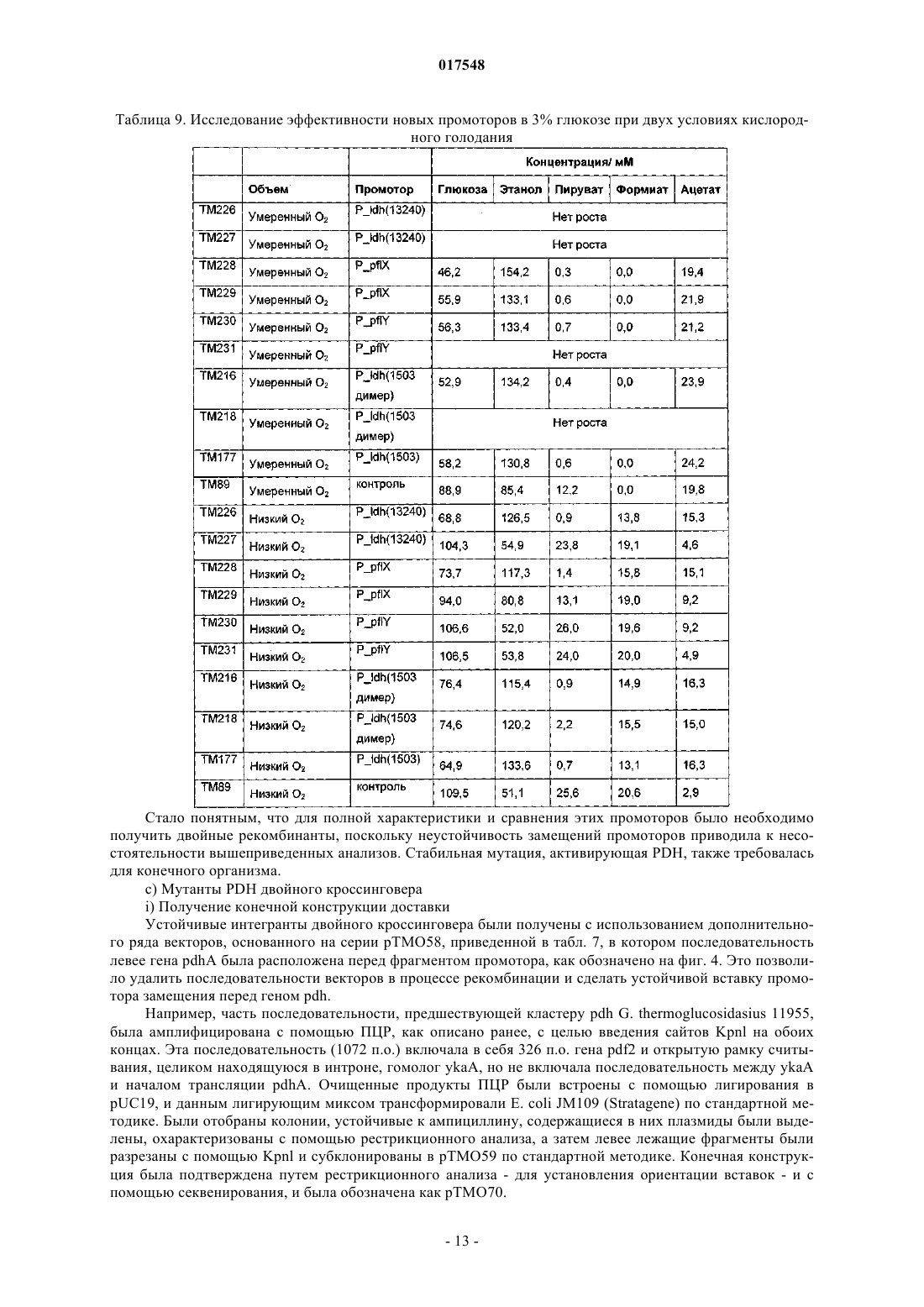
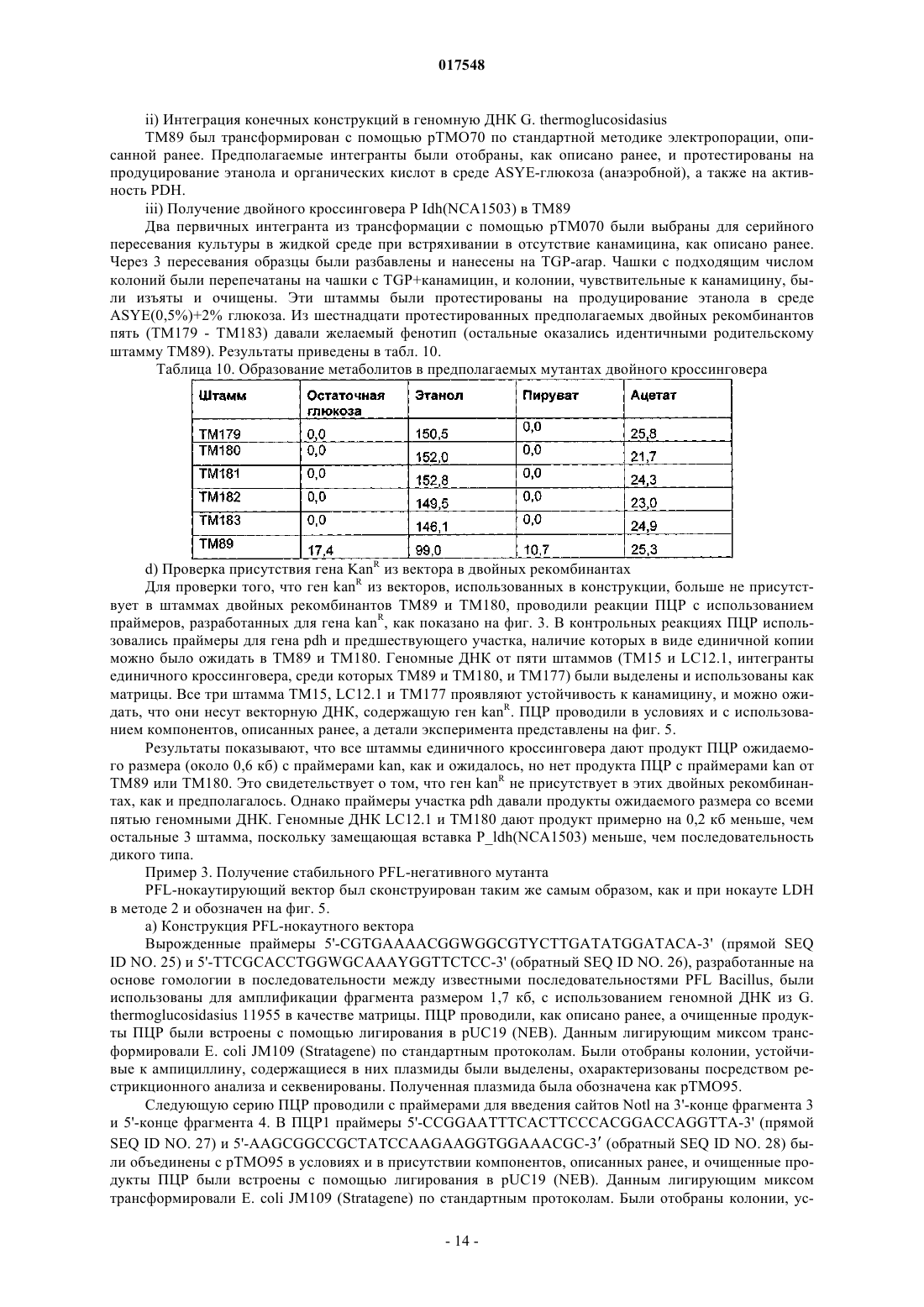
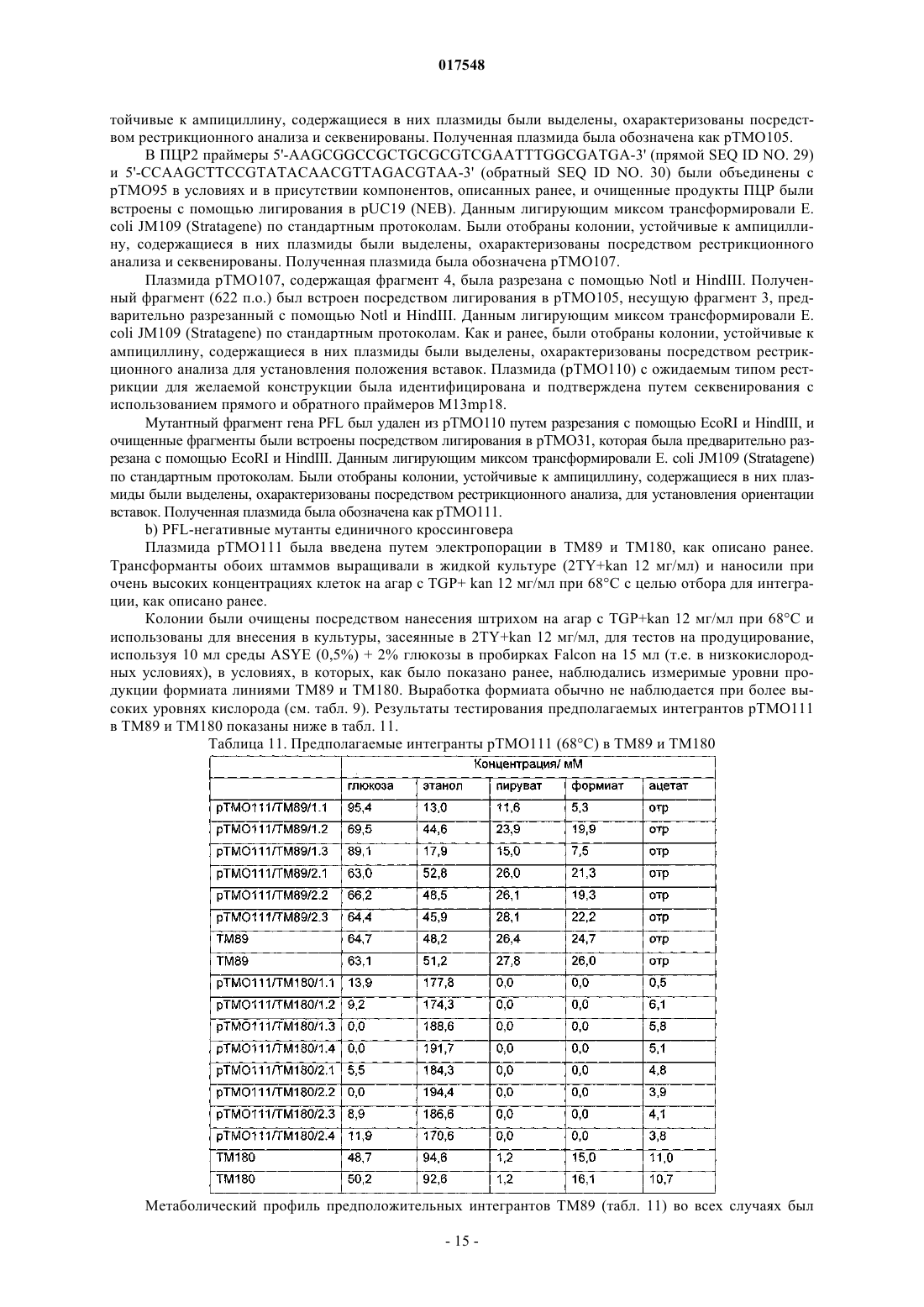
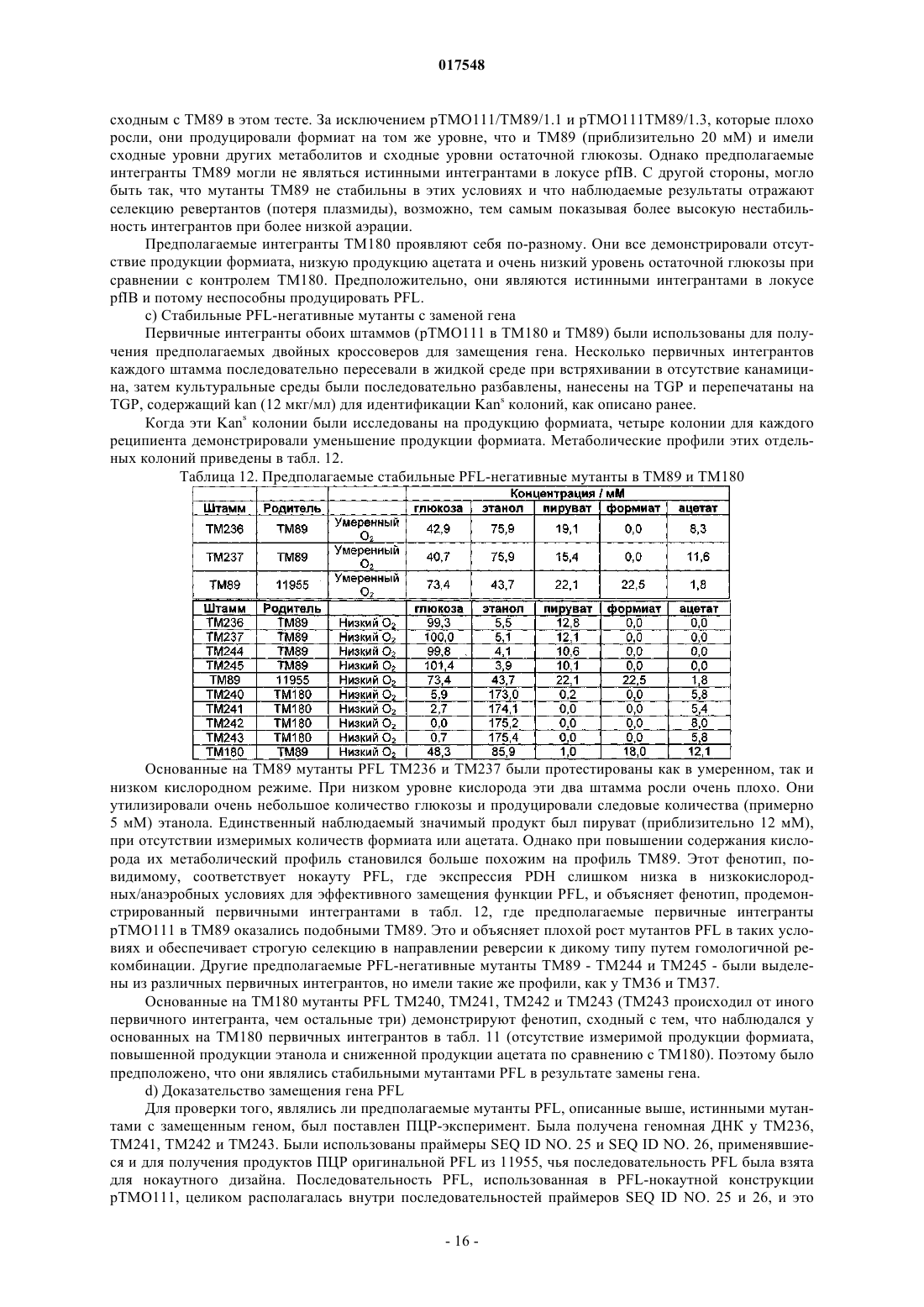
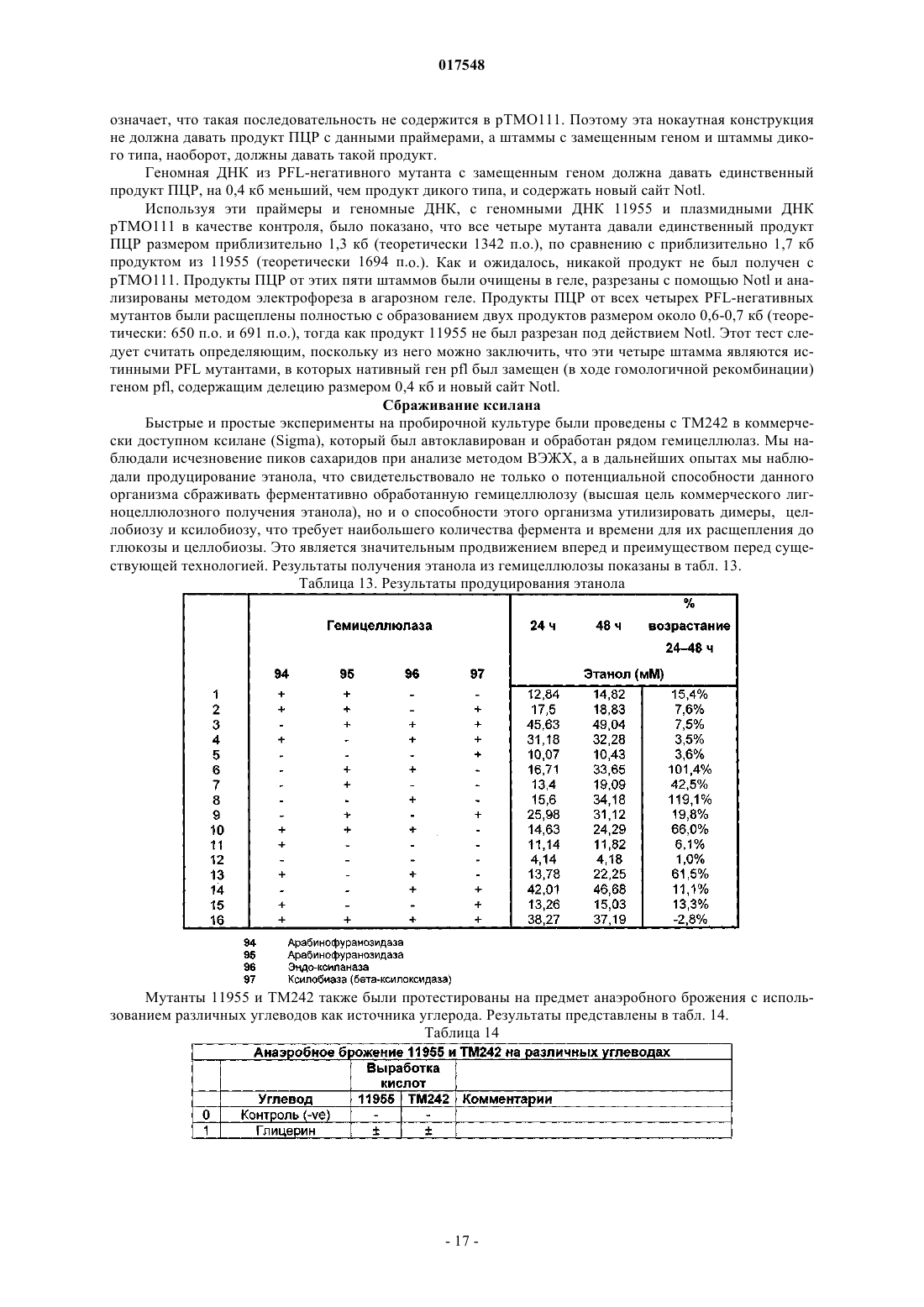
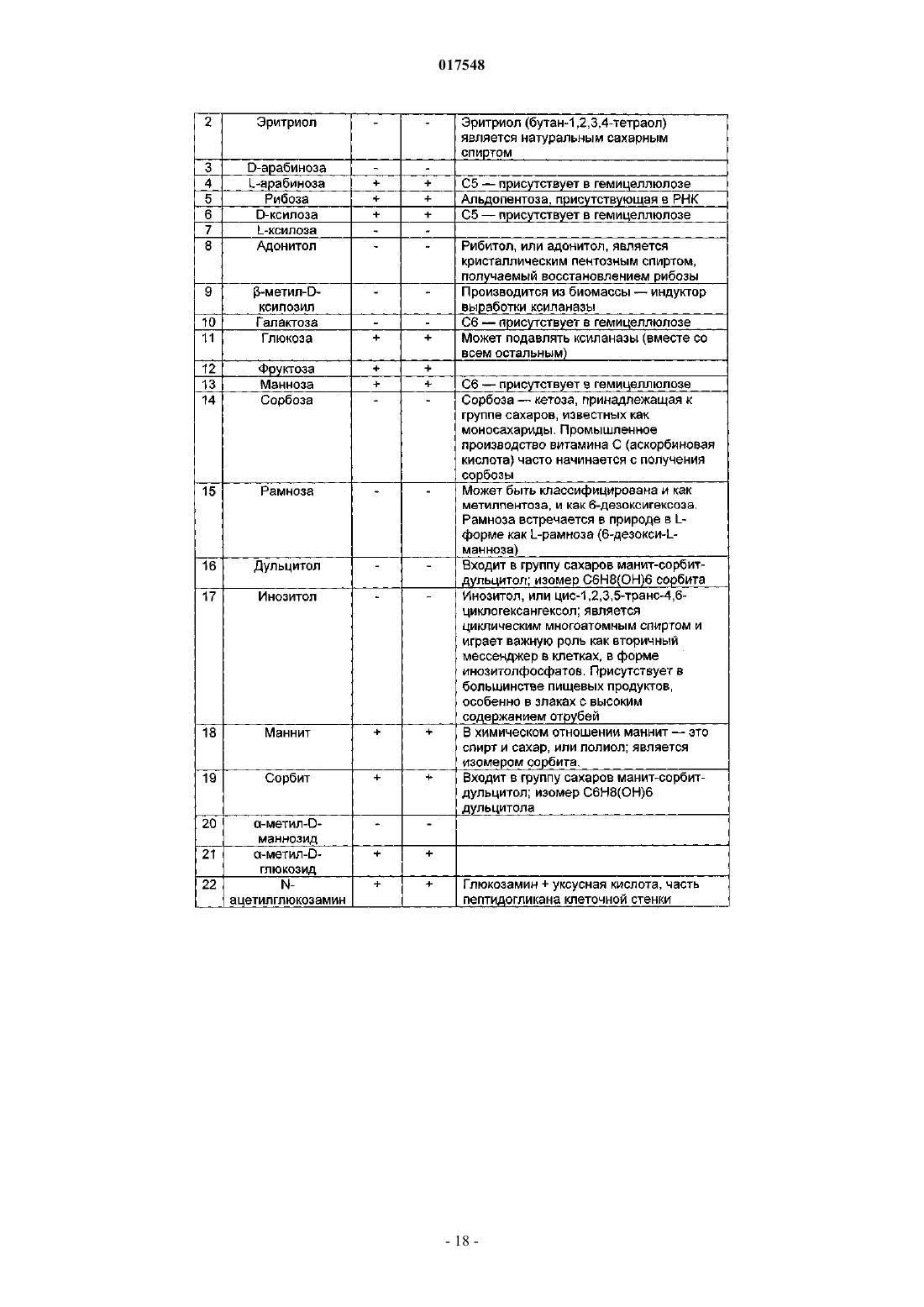
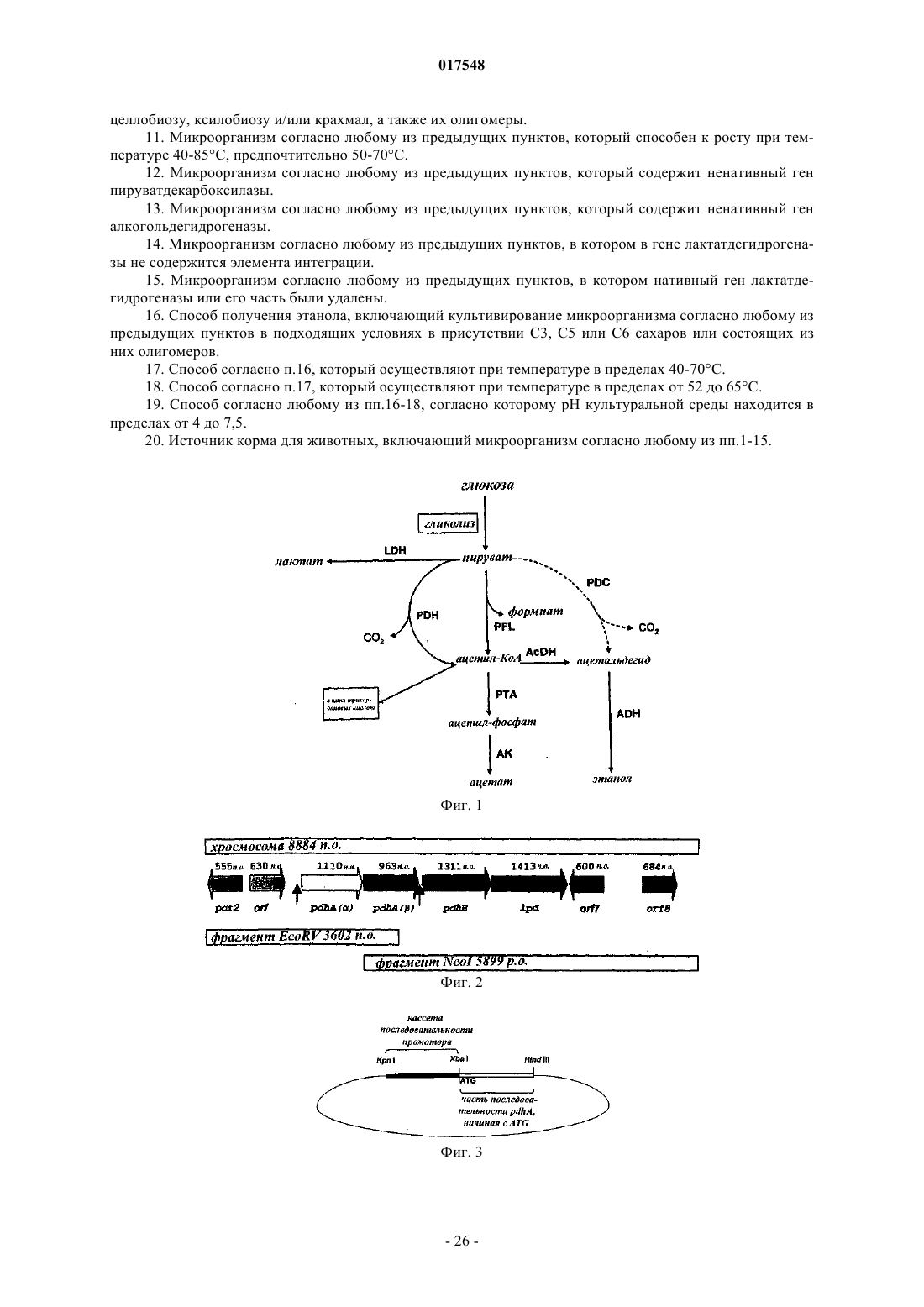
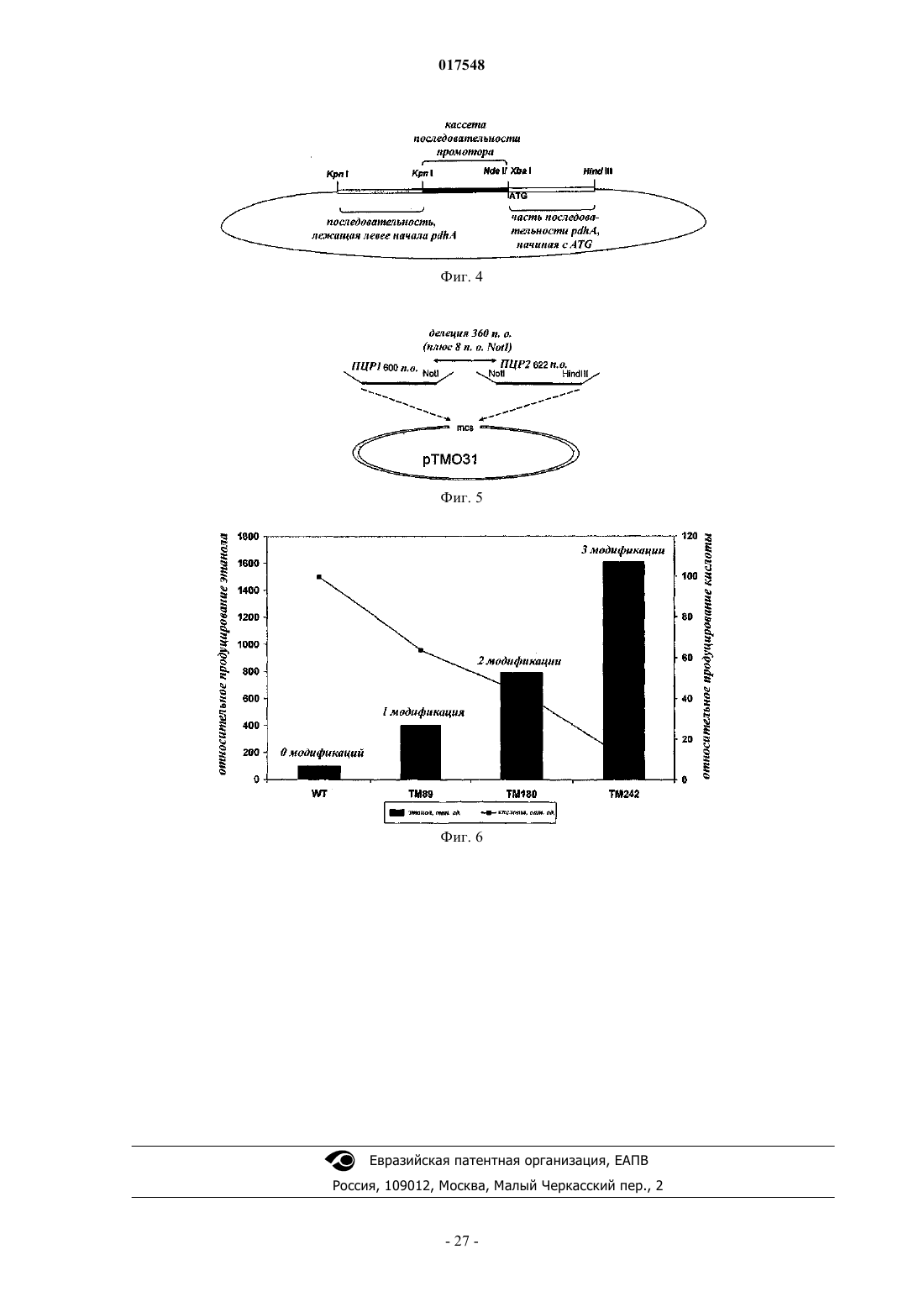
ТЕРМОФИЛЬНЫЙ МИКРООРГАНИЗМ, МОДИФИЦИРОВАННЫЙ ДЛЯ ПОВЫШЕННОЙ ВЫРАБОТКИ ЭТАНОЛА, И СПОСОБ ПОЛУЧЕНИЯ ЭТАНОЛА ПРИ ЕГО ИСПОЛЬЗОВАНИИ Предложен термофильный микроорганизм, модифицированный для повышенной выработки этанола, в котором первая модификация представляет собой инактивацию гена лактатдегидрогеназы, а вторая модификация активирует ген пируватдегидрогеназы.(71)(73) Заявитель и патентовладелец: ТМО РЕНЬЮАБЛЗ ЛИМИТЕД (GB) 017548 Область техники Данное изобретение касается получения этанола как продукта бактериального брожения. В частности, изобретение касается продуцирования этанола термофильными бактериями. Предшествующий уровень техники Бактериальный метаболизм может протекать по различным механизмам, в зависимости от вида бактерий и от условий окружающей среды. Гетеротрофные бактерии, к которым относятся все патогенные микроорганизмы, получают энергию за счет окисления органических соединений; как правило, в роли таких веществ выступают углеводы (особенно глюкоза), липиды и белки. Биологическое окисление этих органических соединений бактериями приводит к синтезу АТФ как источника химической энергии. Этот процесс допускает также продукцию более простых органических веществ (молекулпредшественников), которые требуются бактериальной клетке для биосинтетических реакций. Основной процесс, посредством которого бактерии метаболизируют подходящие субстраты, - это гликолиз, представляющий собой последовательность реакций, в результате которой глюкоза превращается в пируват с одновременным синтезом АТФ. Судьба пирувата в процессе выработки метаболической энергии может быть различной, в зависимости от микроорганизмов и условий окружающей среды. Существуют три основные реакции с участием пирувата. Во-первых, в аэробных условиях многие микроорганизмы получают энергию, используя цикл лимонной кислоты и превращение пирувата в ацетил-коэнзим А, катализируемое пируватдегидрогеназой(PDH). Во-вторых, в анаэробных условиях определенные организмы, продуцирующие этанол, могут осуществлять спиртовое брожение путем декарбоксилирования пирувата до ацетальдегида, которое катализируется пируватдекарбоксилазой (PDC), и последующего восстановления ацетальдегида до этанола с помощью НАДН, которое катализируется алкогольдегидрогеназой (ADH). Третий процесс представляет собой превращение пирувата в лактат, осуществляемое посредством катализа лактатдегидрогеназы (LDH). Большой интерес представляет собой применение микроорганизмов для получения этанола, с использованием как микроорганизмов, естественным образом способных осуществлять анаэробное брожение, так и рекомбинантных микроорганизмов, которые включают в себя гены пируватдекарбоксилазы и алкогольдегидрогеназы. Несмотря на то, что уже имеются некоторые успехи в получении этанола с использованием таких микроорганизмов, процесс брожения часто затрудняется при повышенных концентрациях этанола, особенно если микроорганизмы имеют низкий порог устойчивости к этанолу. Было предложено использовать термофильные бактерии для получения этанола; их применение имеет то преимущество, что брожение можно проводить при повышенных температурах, что позволяет отгонять вырабатываемый этанол в виде пара при температурах свыше 50 С; это позволяет также проводить брожение при высоких концентрациях сахара. Тем не менее, поиск подходящих термофильных бактерий, которые могут эффективно вырабатывать этанол, проблематичен.WO 88/09309 раскрывает продукцию этанола с помощью термофильного штамма Bacillus LLD-R.LLD-R - это неспорулирующий штамм, который возник спонтанно из культуры и в котором ген Idh был инактивирован путем спонтанной мутации или химического мутагенеза. Тем не менее, данный штамм нестабилен, как будет показано ниже.WO 01/49865 раскрывает грамположительную бактерию, которая была трансформирована гетерологичным геном, кодирующим пируватдекарбоксилазу, и которая имеет естественную алкогольдегидрогеназную функцию для выработки этанола. Эта бактерия является термофильной Bacillus, и она может быть модифицирована путем инактивации гена лактатдегидрогеназы посредством вставки транспозона. Бактерии, раскрытые в WO01/49865, выведены из штамма Bacillus LLD-R. Штаммы LN и TN раскрыты как улучшенные производные штамма LLD-R. Однако все штаммы содержат систему рестрикции Hae III типа, которая препятствует трансформации плазмиды и, таким образом, препятствует трансформации в пределах неметилированной ДНК.WO 01/85966 раскрывает микроорганизмы, которые были получены с помощью метилирования invivo, чтобы преодолеть трудности с рестрикцией. Для этого потребовалась трансформация с метилтрансферазой Нае III, полученной из Haemophilus aegyptius, штаммов LLD-R, LN, TN. Однако штаммыLLD-R, LN и TN являются нестабильными мутантами и спонтанно возвращаются к лактатпродуцирующим штаммам дикого типа, особенно при низких рН и высоких концентрациях сахара. Это приводит к изменению продукта брожения с этанола на лактат, что делает данные штаммы непригодными для получения этанола.WO 02/29030 раскрывает, что штамм LLD-R и его производные содержат встречающийся в природе элемент инсерции (insertion element, IE) в кодирующем участке гена Idh. Его траспозиция в ген (или из гена) Idh и последующая инактивация гена нестабильны, что приводит к реверсии. Предложенное решение этой проблемы заключалось в интеграции плазмидной ДНК в последовательность IE. Таким образом, успех получения микроорганизмов для выработки этанола зависит от модификации полученных в лабораторных условиях и химически мутированных микроорганизмов Bacillus, проведения с ними процедуры метилирования in vivo и дальнейшей модификации микроорганизмов с целью инте-1 017548 грации плазмидной ДНК в последовательность IE. Эта процедура сложна, ненадежна, а также имеется нормативное регулирование того, как эти штаммы могут быть использованы. Таким образом, есть необходимость в получении микроорганизмов улучшенного типа, продуцирующих этанол. Сущность изобретения Согласно первому аспекту настоящего изобретения предложен термофильный микроорганизм, модифицированный для повышенной выработки этанола, в котором первая модификация представляет собой инактивацию гена лактатдегидрогеназы, а вторая модификация активирует ген пируватдегидрогеназы. Микроорганизм по изобретению демонстрирует повышенную способность к выработке этанола, по сравнению с диким типом. Согласно второму аспекту настоящего изобретения предложен способ продуцирования этанола,включающий культивирование микроорганизма согласно приведенному выше определению в подходящих условиях в присутствии С 3, С 5 или С 6 сахаров. Краткое описание графических материалов Настоящее изобретение описано со ссылками на сопроводительные графические материалы, где фиг. 1 схематично изображает метаболический путь гликолиза; фиг. 2 изображает гены комплекса пируватдегидрогеназы (PDH); фиг. 3 представляет собой схему конструкции с замещенным промотором для целевой активацииPDH; фиг. 4 представляет собой схему конструкции с замещенным промотором для мутантов двойного кроссовера с активированным геном PDH; фиг. 5 представляет собой схему нокаутной конструкции, предназначенной для получения стабильного PFL-негативного мутанта и, наконец,фиг. 6 является графическим изображением повышенного уровня продукции этанола, который достигается мутантным микроорганизмом по изобретению. Описание изобретения Настоящее изобретение основано на модификации термофильного микроорганизма с целью блокировки экспрессии гена лактатдегидрогеназы и активации гена PDH. Инактивация гена лактатдегидрогеназы позволяет предотвратить расщепление пирувата до лактата и потому способствует (при подходящих условиях) расщеплению пирувата до этанола с помощью пируватдекарбоксилазы и алкогольдегидрогеназы. Предпочтительно, чтобы ген лактатдегидрогеназы был блокирован путем делеции внутри или вне гена. Активация гена PDH способствует превращению пирувата в ацетил-КоА, который далее при подходящих условиях может быть использован для продуцирования ацетальдегида и, в конечном счете, этанола с помощью ацетальдегиддегидрогеназы. Еще одно преимущество активации PDH заключается в том, что уровень пирувата, способного оказывать ингибирующий эффект на захват глюкозы и гликолиз,оказывается сниженным. Это еще сильнее активирует выработку этанола. Для описанной выше модификации можно использовать любой термофильный микроорганизм, но предпочтительно, чтобы он относился к Bacillus spp. Особенно желательно, чтобы это был микроорганизм дикого типа, принадлежащий к видам Geobacillus, а особенно Geobacillus thermoglucosidasius. В предпочтительном воплощении микроорганизмы, выбранные для модификации, называют микроорганизмами дикого типа, т.е. они не являются мутантами, полученными в лабораторных условиях. Микроорганизмы могут быть выделены из образцов окружающей среды, в которых ожидают присутствие термофилов. Выделенные микроорганизмы дикого типа будут способны продуцировать этанол, но в отсутствие модификации основным продуктом брожения, скорее всего, будет лактат. Такие микроорганизмы выбирают также из-за их способности расти на гексозах и/или пентозах, а также на состоящих из них олигомерах, при термофильных температурах. Предпочтительно, чтобы микроорганизм по изобретению имел конкретные желаемые характеристики, которые позволили бы использовать его в процессе брожения. Микроорганизм желательно не должен иметь системы рестрикции, что устранило бы необходимость в метилировании in vivo. Кроме того, микроорганизм должен быть устойчив по меньшей мере в 3% этаноле и должен быть способен усваивать С 3, С 5 и С 6 сахара (или их олигомеры) как субстрат, включая целлобиозу и крахмал. Предпочтительно, чтобы микроорганизм можно было трансформировать с высокой частотой. Более того, микроорганизм должен иметь такую скорость роста в непрерывной культуре, чтобы обеспечивать скорости разбавления порядка 0,3 ч-1 и выше (стандартно 0,3 ОП 600). Микроорганизм должен являться термофилом и быть способным к росту в диапазоне температур 4085 С. Предпочтительно, чтобы микроорганизм был способен к росту в температурном диапазоне 50-70 С. Кроме того, желательно, чтобы микроорганизм мог расти при рН 7,2 и ниже, в частности рН 6,9-рН 4,5. Микроорганизм может быть спорообразующим или может не спорулировать. Успех процесса брожения не обязательно зависит от способности микроорганизма к споруляции, хотя при определенных обстоятельствах может оказаться предпочтительнее работать со спорообразующей бактерией, в случае-2 017548 если хотят использовать микроорганизмы как источник корма для животных по окончании процесса брожения. Это возможно благодаря способности спорулирующих организмов быть хорошими иммунными стимуляторами при их использовании в качестве источника корма для животных. Кроме того, спорообразующие микроорганизмы способны осаждаться в процессе брожения и потому могут быть отделены без помощи центрифугирования. Следовательно, такие микроорганизмы могут использоваться в качестве источника корма для животных, и при этом можно обойтись без сложных или дорогостоящих процедур выделения. Нуклеотидная последовательность для лактатдегидрогеназы теперь известна. Используя эту последовательность, квалифицированный специалист может достичь инактивации гена лактатдегидрогеназы через различные механизмы. Желательно, чтобы ген лактатдегидрогеназы был инактивирован либо путем вставки транспозона, либо, что предпочтительнее, путем делеции последовательности гена или части последовательности гена. Делеция предпочтительнее, поскольку она позволяет избежать проблем с реактивацией последовательности гена, которая часто происходит при использовании инактивации с помощью транспозона. В предпочтительном воплощении ген лактатдегидрогеназы инактивируется путем интеграции температурочувствительной плазмиды (плазмида pUB190-ldh, как описано в PCT/GB06/01586),что приводит к естественной гомологичной рекомбинации или интеграции между плазмидой и хромосомой микроорганизма. Хромосомные интегранты для этой цели могут быть выбраны на основании их устойчивости к антибактериальным агентам (например, канамицину). Интеграция в ген лактатдегидрогеназы может произойти в результате единичной кроссоверной рекомбинации или двух (или более) кроссоверных рекомбинаций. Желательно, чтобы произошел двойной кроссовер, с тем, чтобы удалить ген LDH(или его часть) и изначальный интегрант, т.е. температурочувствительную плазмиду. В этом случае мутантный микроорганизм не будет содержать какой-либо гетерологичной ДНК и потому не будет классифицирован как генетически модифицированный организм (ГМО), согласно положениям о ГМО. Цель второй модификации - активировать PDH. PDH - это большой ферментный комплекс, содержащий три единицы: Е 1 - пируватдекарбоксилаза (ЕС 1.2.4.1, не ЕС 4.1.1.1), Е 2 - дигидролипоамидтрансацетилаза, и Е 3 - дигидролипоамиддегидрогеназа. Для этого комплекса необходим ряд кофакторов,которые включают в себя НАД, ФАД, коэнзим А, липоевую кислоту и тиаминпирофосфат (ТПФ). Этот комплекс кодируется четырьмя генами (поскольку единица Е 1 является гетеродимером, состоящим из субъединици , которые часто обозначаются как pdhA, pdhB, pdhC и pdhD (E1, E1, E2 и Е 3, соответственно). Единица Е 1 пируватдегидрогеназы требует наличия кофермента ТПФ по той же причине,почему и пируватдекарбоксилаза ЕС 4.1.1.1 требует наличия ТПФ и катализирует аналогичную реакцию декарбоксилирования, но в присутствии коэнзима А и липоевой кислоты, переносимыми другими единицами фермента, продуктом является ацетил-КоА, а не ацетальдегид. Однако была зафиксирована пируватдекарбоксилазная активность единицы Е 1, когда последняя была не в комплексе с остальными единицами PDH (LessardPerham; The Journal of Biological Chemistry; 1994, 269:14, 10378-10383; Tomar etal; Applied Microbiology and Biotechnology; 2003, 62, 76-82; Frank et al.; Science; 2004, 306: Oct 29, 872876, supplementary data). Соответственно, пируватдекарбоксилазная активность фермента EC 1.2.4.1 может быть повышена путем активации PDH, с тем, чтобы ацетальдегид продуцировался в большей степени, чем ацетил-КоА. Повышенной пируватдегидрогеназной активности добиваются также с целью устранения узкого места производства, которым является пируват, чтобы обеспечить выработку большего количества этанола при меньшем количестве ацетата и формиата в качестве побочных продуктов. С этой целью были выделены гены PDH и окружающие последовательности с помощью стандартного метода прогулки по хромосоме. Было выделено и секвенировано примерно 8,8 кб ДНК. Было обнаружено, что там содержатся следующие гены, показанные на фиг. 2 и в табл. 1. Гипотетические участки промотора показаны на фиг. 2 (стрелки)один слева от начала pdhA и возможный второй промотор, расположенный перед pdhC. Предыдущий пример вторичного промотора в кластере pdh был описан для Bacillus subtilis (Gao et al.; Journal of Bacteriology, 2002, 184:10, 2780-2788),однако наиболее широко описанные кластеры гена pdh содержат только один промотор слева от кластера(Neveling et al.; Biochimica Acta; 1998 1385, 367-372). Активацию можно проводить с помощью методик,известных в данной области. В частности, активацию можно осуществить путем введения подходящей промоторной или энхансерной последовательности слева от комплекса pdh. Известно, что ферментный комплекс работает как в аэробных, так и в анаэробных условиях (Carlsson et al.; Infection and Immunity; 1985, 49:3, 674-678), но в целом считается, что это аэробный фермент(Ch 15; Principles of Biochemistry; Lehninger, NelsonCox; 2nd Ed, Worth Publishers, New York, 1993,p447), при этом пиурват-формиат-лиаза (PFL) является его анаэробным аналогом. Оба фермента превращают пируват, образовавшийся в ходе гликолиза, в ацетил-KoA, чтобы ввести его в цикл трикарбоновых кислот, но этот цикл работает полностью только в аэробных условиях. Тем не менее, поскольку желательно использовать анаэробные условия, в данном изобретении предпочтительно использовать промоторы, работающие в анаэробных условиях. Таким образом, промоторы для ферментов, которые, как полагают, работают в анаэробных условиях, могут быть промоторы ldh (P ldh из G. stearothermophiliusDSM13240), которые были идентифицированы, выделены и стабильно интегрированы в подходящий участок, левее и в непосредственной близи от начала pdhA. Использованные штаммы и встроенные в комплекс PDH промоторы показаны в табл. 2. В большинстве примеров промоторы дают десятикратное увеличение экспрессии PDH, возрастание захвата глюкозы, уменьшение концентрации пирувата до пренебрежимо малого уровня и 50% возрастание продуцирования этанола. Интересно, что уровень ацетата остается прежним, и это приводит к возрастанию отношения этанол:ацетат в пользу продуцирования этанола. В предпочтительном воплощении третья модификация проводится с целью повышения активностиPDC. Это может быть осуществлено путем инактивации Е 2 (ЕС 2.3.1.12). Инактивацию можно проводить таким же образом, как и инактивацию LDH, но в данном случае мишенью для блокировки выступает ген Е 2. В дальнейшем осуществлении микроорганизм по изобретению включает в себя дальнейшую модификацию, которая инактивирует ген пируват-фармиат-лиазы и таким образом исключает/уменьшает превращение пирувата в ацетил-КоА и формиат. Пируват-формиат-лиаза (PFL) является анаэробным аналогом пируватдегидрогеназы (PDH) и превращает пируват в ацетил-КоА и формиат (см. фиг. 1). Ацетил-КоА может быть превращен в этанол с помощью ацетальдегиддегидрогеназы (AcHD), в то время как формиат представляет собой нежелательный побочный продукт, способный ингибировать рост этанологенных организмов.PFL была выбрана как мишень для нокаутирования, чтобы стимулировать метаболический поток в сторону выработки этанола и улучшить окислительно-восстановительный баланс остального пути синтеза этанола. Дополнительным преимуществом является исключение продукции формиата. Функцию PFL можно инактивировать, используя ту же методику, что и для нокаутирования LDH (описан ниже), при этом получается мутант, который не зависит от выбора антибиотика для поддержания видоизмененного фенотипа. В данном осуществлении, предпочтительно, чтобы микроорганизм претерпел как инактивирование лактатдегидрогеназы, так и активирование пируватдегидрогеназы, с тем чтобы при анаэробных условиях получить повышенную продукцию этанола. Ген PFL может быть инактивирован с использованием методики, описанной для инактивации LDH. Можно использовать инсерцию транспозона, а также генную делецию (или частичную генную делецию). Предпочтительнее использовать генную делецию (или частичную делецию). В дальнейшем предпочтительном воплощении микроорганизм должен также включать в себя гетерологичный ген алкогольдегидрогеназы. Экспрессия такого гетерологичного гена приводит к синтезу ферментов, которые переключают метаболизм таким образом, чтобы этанол был основным продуктом брожения. Этот ген может быть получен от микроорганизмов, естественным образом способных к осуществлению анаэробного брожения, включая виды zymomonas, в т.ч. zymomonas mobilis. Методы получения и внедрения гена в микроорганизмы известны, например, в Ingram et al., BiotechBioEng, 1998; 58 (2+3): 204-214 и US 5916787, содержание этих методов приведено здесь по ссылке. Ген может быть введен в плазмиду или встроен в хромосому по усмотрению квалифицированного специалиста. Микроорганизмы по изобретению можно культивировать в обычных условиях для разведения культуры, в зависимости от выбранного термофильного организма. Выбор субстратов, температуры, рН и других условий роста могут быть выбраны на основании известных требований культуры, например,см. WO 01/49865 и WO 01/85966, содержание каждого из них включено сюда по ссылке.-5 017548 Теперь будет дано описание настоящего изобретения исключительно в виде примера, со ссылками на сопроводительные иллюстрации, в следующих примерах. Инактивация гена лактатдегидрогеназы Пример 1. Получение стабильного мутанта LDH Была разработана стратегия для получения стабильного мутанта гена LDH в Geobacillus thermoglucodasius NCIMB 11955 путем замещения гена, на основании которой возникли два метода. В методе 1 были использованы два существующих уникальных сайта рестрикции вблизи середины последовательности, кодирующей LDH, чтобы произвести делецию. Единственный продукт ПЦР с большой молекулярной массой был получен из геномной ДНК, покрывающей большую часть доступной последовательности LDH, и клонирован в сайт Smal в сайте поликлонирования pUC19 (New England Biolabs). Клон pUC19 затем был последовательно разрезан с помощью BstEII и BsrGI, а затем лигирован,после расщепления фрагментом Кленова, с образованием внутренней делеции в гене LDH между BstEII и BsrGI. В методе 2 ген LDH был клонирован в виде двух продуктов ПЦР, с введением сайтов Notl с показанными ниже олигопраймерами правее, с тем чтобы эти два продукта ПЦР могли соединиться в pUC19,с образованием делеции в середине последовательности ldh. Гены LDH, полученные обоими способами, имеющие внутренние делеции, были субклонированы в три потенциальные системы доставки: pUB190, pNW33N и ТМО 19. В табл. 3 представлены праймеры для ПЦР, использованные во втором способе делеции. Таблица 3 Закрашенные последовательности показывают основания, которые добавлены, чтобы дополнить сайт рестрикции. В табл. 4 перечислены свойства векторов доставки. Таблица 4 Способ 1. Получение геномной ДНК Геномная ДНК была получена из 11955 и использована в качестве матрицы для ПЦР. Клетки из 20 мл ночной культуры 11955 (среда TGP, 52 С) были собраны с помощью центрифугирования (4000 об./мин, 20 мин). Клеточный осадок был ресуспендирован в 5 мл буфера STE (0,3 М сахароза, 25 мМ трис-HCl, 25 мл-6 017548 ЭДТА, рН 8), содержащем 2,5 мг лизоцима и 50 мкл рибонуклеазы А (1 мг/мл). Смесь инкубировали 1 ч при 30 С, после чего добавляли 5 мг протеиназы К и 50 мкл 10% SDS и снова инкубировали 1 ч при 37 С. Лизированную культуру затем последовательно экстрагировали равными объемами смеси фенол:хлороформ (1:1),затем хлороформом и, наконец, осаждали с помощью изопропилового спирта. Промывали дважды 70% ледяным спиртом, после чего осадок ДНК был перерастворен в 0,5 мл буфера ТЕ. Способ 2. Получение конструкций с делецией LDH Проводили ПЦР, используя Robocycler Gradient 96 (Stratagene), условия реакции были следующими: цикл 1 - денатурация при 95 С 5 мин, отжиг при 47 С 1 мин, синтез при 72 С 2 мин, циклы 2-30 денатурация при 95 С 1 мин, отжиг при 47 С 1 мин, синтез при 72 С 2 мин и конечная инкубация при 72 С 5 мин. Использованные ферменты - равная смесь Pfu полимеразы (Promega) и Taq полимеразы (NewEngland Biolabs, NEB). Использованные буферные растворы и нуклеотид-трифосфаты были выбраны согласно инструкции производителя (Pfu, Promega). В качестве матрицы была использована геномная ДНК из 11955, приготовленная вышеуказанным способом, и полученные продукты ПЦР были очищены с помощью электрофореза в агарозном геле, с последующей элюцией из геля, с использованием набора для экстракции из геля (QIAquick Gel extraction kit, Qiagen), согласно инструкции производителя. Очищенные продукты ПЦР были встроены посредством лигирования в pUC19 (NEB), предварительно разрезанную с помощью Smal, и с помощью полученного лигирующего микса трансформировали Escherichiacoli JM109 (Stratagene) по стандартным протоколам. Были отобраны колонии, устойчивые к ампициллину, и содержавшиеся в них плазмиды были выделены и охарактеризованы посредством рестрикционного анализа. Плазмида (рТМО 02) с фрагментом 2, вставленным в pUC19 (с новым сайтом Pstl, введенным на 3'конце фрагмента, и сайтом Notl на 5'-конце, полученными с использованием праймеров 3 и 4), была разрезана посредством Notl и Pstl. Полученный фрагмент (примерно 0,4 кб) был встроен лигированием вpUC19 плазмиду (рТМО 01), несущую фрагмент 1 (полученный с помощью пары праймеров 1 и 2), который также был разрезан с помощью Notl и Pstl. Полученным лигирующим миксом трансформировали Е.coli JM109 (Stratagene) по стандартной методике. Как и ранее, были отобраны колонии, устойчивые к ампициллину, и содержащиеся плазмиды были выделены и охарактеризованы посредством рестрикционного анализа, чтобы установить положение вставок. Плазмида (рТМО 03) с ожидаемым типом рестрикции для желаемой конструкции была идентифицирована и подтверждена с помощью секвенирования с использованием прямого и обратного праймеров М 13mp18. Мутированный фрагмент гена LDH был удален из рТМО 03 посредством расщепления с помощьюHindIII и EcoRI и очищен в ходе электрофореза на агарозном геле, как ранее. Фрагмент был обработан полимеразой Кленова (NEB, согласно инструкциям производителя) с образованием тупых концов для лигирования в pUB190, который был разрезан с помощью Xbal и также обработан полимеразой Кленова. Полученным лигирующим миксом трансформировали E. coli SCS110 (Stratagene) по стандартным протоколам. Как и ранее, были отобраны колонии, устойчивые к ампициллину, и содержащиеся плазмиды были выделены и охарактеризованы посредством рестрикционного анализа. Плазмида (рТМО 14, основанная на скелете pUB190) с ожидаемым типом рестрикции для желаемой конструкции была идентифицирована и использована для трансформации NCIMB 11955 путем электропорации по описанной ниже методике. Методика электропорации для Geobacillus thermoglucosidasius NCIMB 11955 Замороженная исходная культура штамма 11955 была приготовлена путем выращивания ночной культуры в среде TGP (встряхивание при 250 об/мин и 55 С, в объеме 50 мл в конической колбе на 250 мл, ОП 6002), добавления равного объема 20% глицерина, деления на аликвоты по 1 мл и хранения в криопробирках при -80 С. 1 мл этой исходной культуры был использован, чтобы внести его в 50 мл предварительно нагретого TGP в конической колбе на 250 мл, который инкубировали (55 С, 250 об./мин) до достижения значения ОП 600, равного 1,4. Колбу охлаждали на льду в течение 10 мин, затем культуру центрифугировали 20 мин при 4000 об./мин и 4 С в пробирках Falcon на 50 мл. Осадок был ресуспендирован в 50 мл ледяной среды для электропорации и центрифугирован (4000 об./мин, 20 мин). Далее делали три промывки следующим образом: 125 мл и 210 мл, после чего осадок ресуспендировали в 1,5 мл ледяной среды для электропорации,делили на аликвоты 60 мкл и хранили в пробирках Эппендорф на 0,5 мл при -80 С для дальнейшего использования (или использовали сразу). Для электропорации 1-2 мкл ДНК в воде добавляли к 60 мкл электрокомпетентных клеток в пробирке Эппендорф на льду и осторожно перемешивали. Эту суспензию помещали в предварительно охлажденную кювету для электропорации (щель 1 мм) и элетропорировали при 2500 В, емкости 10 мкФ и сопротивлении 600 Ом. Сразу после импульса добавляли 1 мл предварительно нагретого TGP, перемешивали, помещали суспензию в пробирку с завинчивающейся крышкой и инкубировали при 52 С 1 ч на водяной бане с шейкером. После инкубации суспензию либо непосредственно высевали на чашки (например, 20,5 мл), либо центрифугировали (4000 об./мин, 20 мин), ресуспендировали в 200-500 мклTGP, наносили на TGP-агар, содержащий подходящий антибиотик, и выращивали в течение ночи при 52 С. Трансформанты, устойчивые к антибиотику, были видны через 24 ч. Выбор первичных интегрантов для LDH-негативных мутантов единичного кроссовера Плазмиды (например, рТМО 14), использованные для интеграции в геном организма, были температурочувствительными. Основанные на pUB190 нокаутные векторы способны к репликации в организме хозяина при 54 С, но не выше 65 С. Поэтому, чтобы быть способным к росту в присутствии канамицина при 68 С , хозяин должен включить плазмиду в геном. Колония, устойчивая к канамицину, полученная в результате трансформации G. thermoglucodasiusNCIMB 11955 с помощью рТМО 14, была получена путем посева штрихом отдельных колоний на TGPагаре, содержащем 12 мкг/мл канамицина. Этот трансформант был использован для получения первичных интегранотов в 11955 с помощью стимуляции гомологичной рекомбинации с геномным аллелемLDH. Это было достигнуто путем выращивания штамма в 50 мл среды TGP в течение ночи в конической колбе на 250 мл при 52 С и 250 об./мин, центрифугирования культуры (4000 об./мин, 20 мин), ресуспендирования клеток в 1 мл TGP и высевания на чашках с TGP-агаром, содержащим 12 мкг/мл канамицина для последующей инкубации в течение ночи при 68 С. В таких условиях pUB190 не может реплицироваться как автономная плазмида. Большинство колоний, полученных таким образом, при тестировании на продуцирование лактата обнаруживали LDH-фенотип с повышенной способностью вырабатывать этанол, что свидетельствует об образовании LDH-мутантов вследствие интеграции в локусе LDH. Получение мутанта с замещенным геном LDH путем двойного кроссовера Предполагаемый первичный интегрант (ТМ 15) для рТМО 14 был идентифицирован из приведенной выше трансформации и использован для получения двойных рекомбинантов. Это было достигнуто посредством пяти последовательных пересеваний культуры ТМ 15 в среде TGP (5 мл в пробирке falcon на 50 мл, 250 об./мин, 1% пересадка между пересеваемыми культурами) без канамицина, с чередованием условий: 8 ч при 54 С и 16 ч при 52 С. После пяти таких пассажей проводили серийное разведение полученной культуры, и образцы объемом 100 мкл наносили на чашки с TGP и выращивали в течение ночи. Перепечатывание полученных колоний на TGP-агар, содержащий 12 мкг/мл канамицина, было использовано для идентификации колоний, чувствительных к канамицину. После нанесения штрихом отдельных колоний на агар с целью очистки, эти канамицин-чувствительные производные тестировались на продуцирование лактата, и, согласно ожиданиям, они оказались смесью LDH+ и LDH-. Одна LDH- производная, ТМ 89, была далее охарактеризована с помощью ПЦР и Саузерн-блоттинга. Доказательство замены гена LDH Геномная ДНК была получена из ТМ 15 (первичный интегрант) и ТМ 89 (предположительный двойной рекомбинант LDH-) и использована как матрица в ПЦР с использованием праймеров 1 и 4, в описанных выше условиях. Геномная ДНК из 11955 использовалась в качестве контроля. Продукты ПЦР (цепи длиной примерно 0,8 кб были получены из всех трех матриц) были очищены, как описано выше, и образцы были разрезаны с помощью Notl, после чего их анализировали с помощью электрофореза на 0,7% агарозном геле. ПЦР-продукт из 11955 не обнаруживал никаких признаков рестрикции Notl, как и ожидалось, тогда как ПЦР-продукт из ТМ 89 давал 2 полосы примерно в 0,4 кб, что указывало на замещение гена дикого типа мутантным аллелем. Рестрикция Notl ПЦР-продукта из ТМ 15, первичного интегранта,давало 2 линии, видимые и у ТМ 89, со следами неразрезанной (0,8 кб) цепи. Это можно объяснить результатами, полученными при Саузерн-блоттинге геномной ДНК ТМ 15. Геномную ДНК из 11955, ТМ 15 и ТМ 89 разрезали с помощью Notl, Pstl и Notl, а также HindIII иNot, и анализировали с помощью электрофореза на агарозном геле. ДНК помещали на положительно заряженную нейлоновую мембрану (Roche) и гибридизировали с зондом, полученным в ходе ПЦР из гена LDH 11955 с помощью праймеров 1 и 4 (табл. 3), который был помечен DIG (DIG-labeling kit,Roche), согласно инструкциям производителя. Полосы, давшие положительный сигнал после гибридизации с меченым зондом, были выявлены с помощью приложенного набора для детекции (Roche). Результаты Саузерн-блоттинга показали наличие сильно амплифицированной полосы, соответствующей примерно 7,5 кб в ТМ 15, разрезанной посредством Notl, с аналогично амплифицированными полосами около 7,0 и 0,4 кб в ТМ 15, разрезанной посредством HindIII/Notl и Pstl/Notl, соответственно, что свидетель-8 017548 ствует о встраивании множества последовательных копий рТМО 14, интегрированного в локус LDH в этом первичном интегранте. Среди всех трех смесей продуктов расщепления, ТМ 89 продемонстрировал отличный от других тип рестрикции, поскольку давал дополнительную полосу гибридизации, по сравнению с 11955, что согласуется с произошедшим замещением гена. Пример 2. Получение стабильного активированного мутанта PDH а) Клонирование и секвенирование кластера PDH Праймеры 5'-AYGCCCGTTTAAATGRTCGATTTCATG-3' (прямой; SEQ ID NO. 31) и 5'CGAAGTGGCTGGCAATTTGGCTT-3' (обратный; SEQ ID NO. 32) были разработаны на основании гомологии в последовательности между известными последовательностями PDH Bacillus и Geobacillus и были использованы для амплификации фрагмента длиной 1,8 кб с использованием геномной ДНК из G.thermoglucosidasius 11955 в качестве матрицы. ПЦР проводили, как описано ранее, а очищенные продукты ПЦР были лигированы в pUC19 (NEB). Полученным лигирующим миксом трансформировали Е. coliJM109 (Stratagene) по стандартным протоколам. Были отобраны колонии, устойчивые к ампициллину,содержащиеся в них плазмиды были выделены, охарактеризованы с помощью рестрикционного анализа и секвенированы. Первый фрагмент был далее использован как зонд для скрининга последующих библиотек. Геномная ДНК из G. thermoglucosidasius 11955 была расщеплена с помощью 10 различных рестрикционных ферментов по стандартным протоколам. Рестрикционные ферменты BgIII, EcoRV, HindIII иMfel давали фрагменты ДНК размером в пределах 2,5-5 кб, которые были клонированы для формирования нескольких библиотек колоний (pLITMUS28, New England Biolabs, согласно инструкциям производителя). Эти библиотеки были подвергнуты скринингу с помощью меченого зонда ДНК (DIG-labeling kit,Roche, согласно инструкциям производителя). Фрагменты ДНК во всех колониях, которые гибридизовались с зондом, были выделены и секвенированы. Был идентифицирован один клон, который содержал фрагмент геномной ДНК EcoRV длиной 3,6 кб, охватывающий ранее выявленный регион pdh длиной 1,8 кб и продолжающийся ниже от этого региона примерно еще на 1,8 кб. Этот фрагмент кодировал три полных гена, начиная на 5'-конце с пептидной деформилазы 2 (pdf2). Как установлено, правее от pdf2 расположена открытая рамка считывания, кодирующая гипотетический ген (который обнаруживает гомологию с теоретическим белком ykaA(BSU14570) из B. subtilis), тогда как лежащая правее следующая открытая рамка считывания имеет размер 1110 п.о. и, как полагают, кодирует -субъединицу пируватдегидрогеназы А (pdhA(. Непосредственно примыкает к гену pdhA и тянется вплоть до 3'-конца фрагмента EcoRV та часть гена, которая,как полагают, кодирует -субъединицу pdhA (pdhA(. Последовательное расположение генов pdhAисоответствует известным кластерам гена pdh, выявленным у родственных видов. Способ, использованный для идентификации этого фрагмента EcoRV, использовали еще раз для того, чтобы выделить остальную последовательность ДНК. С помощью прямого праймера 5'ACAAGCAAAAGAAGATATTAAAGAG-3' (SEQ ID NO. 5) и обратного праймера 5'TTTAAGTGCTCTAGGAAAATAACAG-3' (SEQ ID NO. 6), которые связываются на 3'-конце фрагментаEcoRV, был получен новый зонд, GT-DIG2, как и ранее, в ходе ПЦР. Ряд рестрикционных ферментов,некоторые из которых, как было показано, разрезают цепь сразу левее от участка GTDIG-2, были использованы для расщепления геномной ДНК, выделенной из G. thermoglucosidasius. Фрагменты, полученные при расщеплении посредством Ncol, были выделены и клонированы, как ранее (pLITMUS28, New England Biolabs, согласно инструкциям производителя). В ходе скрининга с помощью зонда GT-DIG2 был идентифицирован клон, содержащий фрагмент ДНК Ncol, состоящий из ДНК длиной 6 кб, расположенный правее фрагмента EcoRV. Последовательности фрагментов Ncol иEcoRV образовывали единую непрерывную последовательность (контиг) длиной 8884 п.о. Анализ последовательности выявил, что этот контиг вмещает кластер гена pdh, состоящий из четырех смежных генов, зажатых с обеих сторон между двумя гипотетическими генами. Организация генов, закодированных внутри контига, показана на фиг. 2.b) Мутанты PDH единичного кроссовера - доказательство концепции Стратегия, примененная для получения активированных мутантных генов PDH в LDH- мутантах G.thermoglucosidasius 11955, включала использование ранее идентифицированной последовательностиpdhA для разработки и получения кассеты с кодирующей последовательностью pdhA с предшествующим ей сайтом рестрикции, который обеспечивает легкость вставки гетерологичных промоторов, и последующую их вставку в подходящий вектор интеграции. Требовался сильный промотор, такой как LDHпромотор из G. thermoglucosidasius или G. steareothermophilus. Суть метода отображена на фиг. 3. Требуемые мутанты были получены путем трансформации интегрирующего вектора в штамм хозяина и отбора интегрантов, устойчивых к канамицину. Подобные мутанты единичного кроссовера неустойчивы в отсутствие антибиотика, так как процесс интеграции легко обратим.i) Разработка векторов для проведения активации Для того чтобы описанная выше стратегия работала, нужно было сконструировать новые векторы доставки, чтобы могли быть использованы Ndel-сайты рестрикции внутри сайтов поликлонированияpUB190. Таблица 5. Векторы, разработанные для проведения активацииii) фрагмент скелета pdhA Описанная ранее последовательность pdhk, полученная из исходного продукта ПЦР размером 1,8 кб и клонированная в pUC19, была использована для разработки прямого праймера 5'AATCTAGACATATGGGTGCGAAAACATCCAGATT-3' (SEQ ID NO. 7), содержащего сайты Xbal/Ndel,так что концевой фрагмент ATG в сайте Ndel предположительно представлял собой стартовый кодонATG гена pdhA. Этот стартовый кодон был определен при выверке расположения остальных генов pdhA и тестировании возможных рамок считывания. Этот праймер был использован для конъюгации с обратным праймером 5'CCAAGCTTTCTTTAATATCTTCTTTTGCTTG-3' (SEQ ID NO. 8) (включающим сайт HindIII) для амплификации передней части гена pdhA, по описанной выше методике проведения ПЦР. Был получен продукт ПЦР размером около 1 кб, который затем очищали и встраивали с помощью лигирования вpUC19 (NEB). Данным лигирующим миксом трансформировали Е. coli JM109 (Stratagene) по стандартной методике. Были отобраны колонии, устойчивые к ампициллину, содержащиеся в них плазмиды были выделены, охарактеризованы с помощью рестрикционного анализа, а затем фрагмент pdhA был разрезан с помощью Ndel и HindIII и субклонирован в рТМО 31 по стандартной методике. Конечная конструкция в рТМО 31 была подтверждена путем секвенирования и была обозначена как рТМО 46.iii) Фрагменты промоторов и получение конечной конструкции доставки Фрагменты промоторов были затем клонированы в конструкцию рТМО 46 - предшествующую генуpdhA - как фрагменты Kpnl/Ndel. Для исходных конструкций были выбраны следующие фрагменты промоторов.- 10017548 Таблица 6. Промоторы, использованные для активации PDH, и их источники Были разработаны праймеры для получения этих промотрных участков из геномной ДНК (выделенной из G. thermoglucosidasius 11955, G. stearothermophilus NCA1503, G. stearothermophilus DSM13240 и В. cereus ATCC14579, как описано ранее), как фрагментов Kpnl/Ndel, и были получены продукты ПЦР с использованием описанных ранее компонентов и методик. Очищенные продукты ПЦР были встроены с помощью лигирования в рТМО 23 (pUC19 с исключенным сайтом Ndel), и данным лигирующим миксом трансформировали Е. colli JM109 (Stratagene) по стандартной методике. Были отобраны колонии, устойчивые к ампициллину, содержащиеся в них плазмиды были выделены, охарактеризованы с помощью рестрикционного анализа, а затем фрагменты промоторов были разрезаны с помощью Ndel и HindIII и субклонированы в рТМО 46 по стандартной методике. Конечные конструкции в рТМО 46 были подтверждены путем секвенирования и были обозначены как плазмиды в табл. 7.- 11017548 Таблица 7. Конструкции одинарного кроссовера для активации PDHiv) Интеграция конечных конструкций в геномную ДНК G. thermoglucosidasius ТМ 89 были трансформированы приведенными выше плазмидами по стандартной методике электропорации, описанной ранее. Предполагаемые интегранты были отобраны, как описано ранее, и протестированы на предмет продуцирования этанола и органических кислот в глюкозной ASYE среде (анаэробной), а также на пируватдегидрогеназную активность. Результаты показаны в табл. 8. Таблица 8. Анализ на предмет PDH новых промоторных интегрантов на 8-часовых культурах в среде Как оказалось, новые промоторы давали сравнительно высокие уровни активности PDH, значительно выше, чем ТМ 89 в данной точке времени (однако низкий уровень белка, полученный для ТМ 89, свидетельствует, что ТМ 89, возможно, миновал свою точку пика продукции). Слабое соответствие между этими двумя интегрантами каждого промотора делает затруднительным сравнение относительной силы этих промоторов. Те же штаммы, что тестировались на уровень PDH, были также протестированы на продуцирование этанола при повышенном уровне глюкозы и низкой аэрации, в дальнейшей попытке установить, имеют ли хотя бы некоторые из новых промоторов какие-либо преимущества перед оригинальными промоторами. Результаты представлены ниже в табл. 9.- 12017548 Таблица 9. Исследование эффективности новых промоторов в 3% глюкозе при двух условиях кислородного голодания Стало понятным, что для полной характеристики и сравнения этих промоторов было необходимо получить двойные рекомбинанты, поскольку неустойчивость замещений промоторов приводила к несостоятельности вышеприведенных анализов. Стабильная мутация, активирующая PDH, также требовалась для конечного организма. с) Мутанты PDH двойного кроссинговераi) Получение конечной конструкции доставки Устойчивые интегранты двойного кроссинговера были получены с использованием дополнительного ряда векторов, основанного на серии рТМО 58, приведенной в табл. 7, в котором последовательность левее гена pdhA была расположена перед фрагментом промотора, как обозначено на фиг. 4. Это позволило удалить последовательности векторов в процессе рекомбинации и сделать устойчивой вставку промотора замещения перед геном pdh. Например, часть последовательности, предшествующей кластеру pdh G. thermoglucosidasius 11955,была амплифицирована с помощью ПЦР, как описано ранее, с целью введения сайтов Kpnl на обоих концах. Эта последовательность (1072 п.о.) включала в себя 326 п.о. гена pdf2 и открытую рамку считывания, целиком находящуюся в интроне, гомолог ykaA, но не включала последовательность между ykaA и началом трансляции pdhA. Очищенные продукты ПЦР были встроены с помощью лигирования вpUC19, и данным лигирующим миксом трансформировали E. coli JM109 (Stratagene) по стандартной методике. Были отобраны колонии, устойчивые к ампициллину, содержащиеся в них плазмиды были выделены, охарактеризованы с помощью рестрикционного анализа, а затем левее лежащие фрагменты были разрезаны с помощью Kpnl и субклонированы в рТМО 59 по стандартной методике. Конечная конструкция была подтверждена путем рестрикционного анализа - для установления ориентации вставок - и с помощью секвенирования, и была обозначена как рТМО 70.ii) Интеграция конечных конструкций в геномную ДНК G. thermoglucosidasius ТМ 89 был трансформирован с помощью рТМО 70 по стандартной методике электропорации, описанной ранее. Предполагаемые интегранты были отобраны, как описано ранее, и протестированы на продуцирование этанола и органических кислот в среде ASYE-глюкоза (анаэробной), а также на активность PDH.iii) Получение двойного кроссинговера Р Idh(NCA1503) в ТМ 89 Два первичных интегранта из трансформации с помощью рТМ 070 были выбраны для серийного пересевания культуры в жидкой среде при встряхивании в отсутствие канамицина, как описано ранее. Через 3 пересевания образцы были разбавлены и нанесены на TGP-arap. Чашки с подходящим числом колоний были перепечатаны на чашки с TGP+канамицин, и колонии, чувствительные к канамицину, были изъяты и очищены. Эти штаммы были протестированы на продуцирование этанола в средеASYE(0,5%)+2% глюкоза. Из шестнадцати протестированных предполагаемых двойных рекомбинантов пять (ТМ 179 - ТМ 183) давали желаемый фенотип (остальные оказались идентичными родительскому штамму ТМ 89). Результаты приведены в табл. 10. Таблица 10. Образование метаболитов в предполагаемых мутантах двойного кроссинговераd) Проверка присутствия гена KanR из вектора в двойных рекомбинантах Для проверки того, что ген kanR из векторов, использованных в конструкции, больше не присутствует в штаммах двойных рекомбинантов ТМ 89 и ТМ 180, проводили реакции ПЦР с использованием праймеров, разработанных для гена kanR, как показано на фиг. 3. В контрольных реакциях ПЦР использовались праймеры для гена pdh и предшествующего участка, наличие которых в виде единичной копии можно было ожидать в ТМ 89 и ТМ 180. Геномные ДНК от пяти штаммов (ТМ 15 и LC12.1, интегранты единичного кроссинговера, среди которых ТМ 89 и ТМ 180, и ТМ 177) были выделены и использованы как матрицы. Все три штамма ТМ 15, LC12.1 и ТМ 177 проявляют устойчивость к канамицину, и можно ожидать, что они несут векторную ДНК, содержащую ген kanR. ПЦР проводили в условиях и с использованием компонентов, описанных ранее, а детали эксперимента представлены на фиг. 5. Результаты показывают, что все штаммы единичного кроссинговера дают продукт ПЦР ожидаемого размера (около 0,6 кб) с праймерами kаn, как и ожидалось, но нет продукта ПЦР с праймерами kаn от ТМ 89 или ТМ 180. Это свидетельствует о том, что ген kanR не присутствует в этих двойных рекомбинантах, как и предполагалось. Однако праймеры участка pdh давали продукты ожидаемого размера со всеми пятью геномными ДНК. Геномные ДНК LC12.1 и ТМ 180 дают продукт примерно на 0,2 кб меньше, чем остальные 3 штамма, поскольку замещающая вставка Pldh(NCA1503) меньше, чем последовательность дикого типа. Пример 3. Получение стабильного PFL-негативного мутантаPFL-нокаутирующий вектор был сконструирован таким же самым образом, как и при нокауте LDH в методе 2 и обозначен на фиг. 5. а) Конструкция PFL-нокаутного вектора Вырожденные праймеры 5'-CGTGAAAACGGWGGCGTYCTTGATATGGATACA-3' (прямой SEQID NO. 25) и 5'-TTCGCACCTGGWGCAAAYGGTTCTCC-3' (обратный SEQ ID NO. 26), разработанные на основе гомологии в последовательности между известными последовательностями PFL Bacillus, были использованы для амплификации фрагмента размером 1,7 кб, с использованием геномной ДНК из G.thermoglucosidasius 11955 в качестве матрицы. ПЦР проводили, как описано ранее, а очищенные продукты ПЦР были встроены с помощью лигирования в pUC19 (NEB). Данным лигирующим миксом трансформировали Е. coli JM109 (Stratagene) по стандартным протоколам. Были отобраны колонии, устойчивые к ампициллину, содержащиеся в них плазмиды были выделены, охарактеризованы посредством рестрикционного анализа и секвенированы. Полученная плазмида была обозначена как рТМО 95. Следующую серию ПЦР проводили с праймерами для введения сайтов Notl на 3'-конце фрагмента 3 и 5'-конце фрагмента 4. В ПЦР 1 праймеры 5'-CCGGAATTTCACTTCCCACGGACCAGGTTA-3' (прямойSEQ ID NO. 27) и 5'-AAGCGGCCGCTATCCAAGAAGGTGGAAACGC-3 (обратный SEQ ID NO. 28) были объединены с рТМО 95 в условиях и в присутствии компонентов, описанных ранее, и очищенные продукты ПЦР были встроены с помощью лигирования в pUC19 (NEB). Данным лигирующим миксом трансформировали Е. coli JM109 (Stratagene) по стандартным протоколам. Были отобраны колонии, ус- 14017548 тойчивые к ампициллину, содержащиеся в них плазмиды были выделены, охарактеризованы посредством рестрикционного анализа и секвенированы. Полученная плазмида была обозначена как рТМО 105. В ПЦР 2 праймеры 5'-AAGCGGCCGCTGCGCGTCGAATTTGGCGATGA-3' (прямой SEQ ID NO. 29) и 5'-CCAAGCTTCCGTATACAACGTTAGACGTAA-3' (обратный SEQ ID NO. 30) были объединены с рТМО 95 в условиях и в присутствии компонентов, описанных ранее, и очищенные продукты ПЦР были встроены с помощью лигирования в pUC19 (NEB). Данным лигирующим миксом трансформировали Е.coli JM109 (Stratagene) по стандартным протоколам. Были отобраны колонии, устойчивые к ампициллину, содержащиеся в них плазмиды были выделены, охарактеризованы посредством рестрикционного анализа и секвенированы. Полученная плазмида была обозначена рТМО 107. Плазмида рТМО 107, содержащая фрагмент 4, была разрезана с помощью Notl и HindIII. Полученный фрагмент (622 п.о.) был встроен посредством лигирования в pTMO105, несущую фрагмент 3, предварительно разрезанный с помощью Notl и HindIII. Данным лигирующим миксом трансформировали E.coli JM109 (Stratagene) по стандартным протоколам. Как и ранее, были отобраны колонии, устойчивые к ампициллину, содержащиеся в них плазмиды были выделены, охарактеризованы посредством рестрикционного анализа для установления положения вставок. Плазмида (рТМО 110) с ожидаемым типом рестрикции для желаемой конструкции была идентифицирована и подтверждена путем секвенирования с использованием прямого и обратного праймеров M13mp18. Мутантный фрагмент гена PFL был удален из рТМО 110 путем разрезания с помощью EcoRI и HindIII, и очищенные фрагменты были встроены посредством лигирования в рТМО 31, которая была предварительно разрезана с помощью EcoRI и HindIII. Данным лигирующим миксом трансформировали Е. coli JM109 (Stratagene) по стандартным протоколам. Были отобраны колонии, устойчивые к ампициллину, содержащиеся в них плазмиды были выделены, охарактеризованы посредством рестрикционного анализа, для установления ориентации вставок. Полученная плазмида была обозначена как рТМО 111.b) PFL-негативные мутанты единичного кроссинговера Плазмида рТМО 111 была введена путем электропорации в ТМ 89 и ТМ 180, как описано ранее. Трансформанты обоих штаммов выращивали в жидкой культуре (2TY+kan 12 мг/мл) и наносили при очень высоких концентрациях клеток на агар с TGP+ kan 12 мг/мл при 68 С с целью отбора для интеграции, как описано ранее. Колонии были очищены посредством нанесения штрихом на агар с TGP+kan 12 мг/мл при 68 С и использованы для внесения в культуры, засеянные в 2TY+kan 12 мг/мл, для тестов на продуцирование,используя 10 мл среды ASYE (0,5%) + 2% глюкозы в пробирках Falcon на 15 мл (т.е. в низкокислородных условиях), в условиях, в которых, как было показано ранее, наблюдались измеримые уровни продукции формиата линиями ТМ 89 и ТМ 180. Выработка формиата обычно не наблюдается при более высоких уровнях кислорода (см. табл. 9). Результаты тестирования предполагаемых интегрантов рТМО 111 в ТМ 89 и ТМ 180 показаны ниже в табл. 11. Таблица 11. Предполагаемые интегранты рТМО 111 (68 С) в ТМ 89 и ТМ 180 Метаболический профиль предположительных интегрантов ТМ 89 (табл. 11) во всех случаях был- 15017548 сходным с ТМ 89 в этом тесте. За исключением рТМО 111/ТМ 89/1.1 и рТМО 111 ТМ 89/1.3, которые плохо росли, они продуцировали формиат на том же уровне, что и ТМ 89 (приблизительно 20 мМ) и имели сходные уровни других метаболитов и сходные уровни остаточной глюкозы. Однако предполагаемые интегранты ТМ 89 могли не являться истинными интегрантами в локусе pfIB. С другой стороны, могло быть так, что мутанты ТМ 89 не стабильны в этих условиях и что наблюдаемые результаты отражают селекцию ревертантов (потеря плазмиды), возможно, тем самым показывая более высокую нестабильность интегрантов при более низкой аэрации. Предполагаемые интегранты ТМ 180 проявляют себя по-разному. Они все демонстрировали отсутствие продукции формиата, низкую продукцию ацетата и очень низкий уровень остаточной глюкозы при сравнении с контролем ТМ 180. Предположительно, они являются истинными интегрантами в локусеpfIB и потому неспособны продуцировать PFL. с) Стабильные PFL-негативные мутанты с заменой гена Первичные интегранты обоих штаммов (рТМО 111 в ТМ 180 и ТМ 89) были использованы для получения предполагаемых двойных кроссоверов для замещения гена. Несколько первичных интегрантов каждого штамма последовательно пересевали в жидкой среде при встряхивании в отсутствие канамицина, затем культуральные среды были последовательно разбавлены, нанесены на TGP и перепечатаны наTGP, содержащий kan (12 мкг/мл) для идентификации Kans колоний, как описано ранее. Когда эти Kans колонии были исследованы на продукцию формиата, четыре колонии для каждого реципиента демонстрировали уменьшение продукции формиата. Метаболические профили этих отдельных колоний приведены в табл. 12. Таблица 12. Предполагаемые стабильные PFL-негативные мутанты в ТМ 89 и ТМ 180 Основанные на ТМ 89 мутанты PFL TM236 и ТМ 237 были протестированы как в умеренном, так и низком кислородном режиме. При низком уровне кислорода эти два штамма росли очень плохо. Они утилизировали очень небольшое количество глюкозы и продуцировали следовые количества (примерно 5 мМ) этанола. Единственный наблюдаемый значимый продукт был пируват (приблизительно 12 мМ),при отсутствии измеримых количеств формиата или ацетата. Однако при повышении содержания кислорода их метаболический профиль становился больше похожим на профиль ТМ 89. Этот фенотип, повидимому, соответствует нокауту PFL, где экспрессия PDH слишком низка в низкокислородных/анаэробных условиях для эффективного замещения функции PFL, и объясняет фенотип, продемонстрированный первичными интегрантами в табл. 12, где предполагаемые первичные интегранты рТМО 111 в ТМ 89 оказались подобными ТМ 89. Это и объясняет плохой рост мутантов PFL в таких условиях и обеспечивает строгую селекцию в направлении реверсии к дикому типу путем гомологичной рекомбинации. Другие предполагаемые PFL-негативные мутанты ТМ 89 - ТМ 244 и ТМ 245 - были выделены из различных первичных интегрантов, но имели такие же профили, как у ТМ 36 и ТМ 37. Основанные на ТМ 180 мутанты PFL ТМ 240, ТМ 241, ТМ 242 и ТМ 243 (ТМ 243 происходил от иного первичного интегранта, чем остальные три) демонстрируют фенотип, сходный с тем, что наблюдался у основанных на ТМ 180 первичных интегрантов в табл. 11 (отсутствие измеримой продукции формиата,повышенной продукции этанола и сниженной продукции ацетата по сравнению с ТМ 180). Поэтому было предположено, что они являлись стабильными мутантами PFL в результате замены гена.d) Доказательство замещения гена PFL Для проверки того, являлись ли предполагаемые мутанты PFL, описанные выше, истинными мутантами с замещенным геном, был поставлен ПЦР-эксперимент. Была получена геномная ДНК у ТМ 236,ТМ 241, ТМ 242 и ТМ 243. Были использованы праймеры SEQ ID NO. 25 и SEQ ID NO. 26, применявшиеся и для получения продуктов ПЦР оригинальной PFL из 11955, чья последовательность PFL была взята для нокаутного дизайна. Последовательность PFL, использованная в PFL-нокаутной конструкции рТМО 111, целиком располагалась внутри последовательностей праймеров SEQ ID NO. 25 и 26, и это- 16017548 означает, что такая последовательность не содержится в рТМО 111. Поэтому эта нокаутная конструкция не должна давать продукт ПЦР с данными праймерами, а штаммы с замещенным геном и штаммы дикого типа, наоборот, должны давать такой продукт. Геномная ДНК из PFL-негативного мутанта с замещенным геном должна давать единственный продукт ПЦР, на 0,4 кб меньший, чем продукт дикого типа, и содержать новый сайт Notl. Используя эти праймеры и геномные ДНК, с геномными ДНК 11955 и плазмидными ДНК рТМО 111 в качестве контроля, было показано, что все четыре мутанта давали единственный продукт ПЦР размером приблизительно 1,3 кб (теоретически 1342 п.о.), по сравнению с приблизительно 1,7 кб продуктом из 11955 (теоретически 1694 п.о.). Как и ожидалось, никакой продукт не был получен с рТМО 111. Продукты ПЦР от этих пяти штаммов были очищены в геле, разрезаны с помощью Notl и анализированы методом электрофореза в агарозном геле. Продукты ПЦР от всех четырех PFL-негативных мутантов были расщеплены полностью с образованием двух продуктов размером около 0,6-0,7 кб (теоретически: 650 п.о. и 691 п.о.), тогда как продукт 11955 не был разрезан под действием Notl. Этот тест следует считать определяющим, поскольку из него можно заключить, что эти четыре штамма являются истинными PFL мутантами, в которых нативный ген pfl был замещен (в ходе гомологичной рекомбинации) геном pfl, содержащим делецию размером 0,4 кб и новый сайт Notl. Сбраживание ксилана Быстрые и простые эксперименты на пробирочной культуре были проведены с ТМ 242 в коммерчески доступном ксилане (Sigma), который был автоклавирован и обработан рядом гемицеллюлаз. Мы наблюдали исчезновение пиков сахаридов при анализе методом ВЭЖХ, а в дальнейших опытах мы наблюдали продуцирование этанола, что свидетельствовало не только о потенциальной способности данного организма сбраживать ферментативно обработанную гемицеллюлозу (высшая цель коммерческого лигноцеллюлозного получения этанола), но и о способности этого организма утилизировать димеры, целлобиозу и ксилобиозу, что требует наибольшего количества фермента и времени для их расщепления до глюкозы и целлобиозы. Это является значительным продвижением вперед и преимуществом перед существующей технологией. Результаты получения этанола из гемицеллюлозы показаны в табл. 13. Таблица 13. Результаты продуцирования этанола Мутанты 11955 и ТМ 242 также были протестированы на предмет анаэробного брожения с использованием различных углеводов как источника углерода. Результаты представлены в табл. 14. Таблица 14 Микроорганизм, обозначенный здесь как ТМ 89, и плазмида pUB 190-Idh были депонированы вNCIMB под номерами доступа 41275 и 41276, соответственно. Депозитарий: NCIMB Ltd, Ferguson Building, Craibstone Estate, Bucksburn, Aberdeen, AB21 9YA, United Kingdom. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Термофильный микроорганизм, модифицированный для повышенной выработки этанола, в котором первая модификация представляет собой инактивацию гена лактатдегидрогеназы, а вторая модификация активирует ген пируватдегидрогеназы. 2. Микроорганизм согласно п.1, в котором вторая модификация представляет собой вставку промотора гена левее гена пируватдегидрогеназы, причем промотор работает в анаэробных условиях. 3. Микроорганизм согласно п.1 или 2, дополнительно модифицированный с целью инактивации гена дигидролипоамид-трансацетилазы. 4. Микроорганизм согласно любому из предыдущих пунктов, дополнительно модифицированный с целью инактивации гена пируватформиатлиазы. 5. Микроорганизм согласно любому из предыдущих пунктов, не содержащий системы рестрикции. 6. Микроорганизм согласно любому из предыдущих пунктов, который является спорообразующим. 7. Микроорганизм согласно любому из предыдущих пунктов, который относится к виду Geobacillus. 8. Микроорганизм по п.7, который представляет собой Geobacillus thermoglucosidasius. 9. Микроорганизм согласно любому из предыдущих пунктов, который стабилен в культуральной среде с содержанием этанола вплоть до 30% (вес./об.). 10. Микроорганизм согласно любому из предыдущих пунктов, который способен метаболизировать- 25017548 целлобиозу, ксилобиозу и/или крахмал, а также их олигомеры. 11. Микроорганизм согласно любому из предыдущих пунктов, который способен к росту при температуре 40-85 С, предпочтительно 50-70 С. 12. Микроорганизм согласно любому из предыдущих пунктов, который содержит ненативный ген пируватдекарбоксилазы. 13. Микроорганизм согласно любому из предыдущих пунктов, который содержит ненативный ген алкогольдегидрогеназы. 14. Микроорганизм согласно любому из предыдущих пунктов, в котором в гене лактатдегидрогеназы не содержится элемента интеграции. 15. Микроорганизм согласно любому из предыдущих пунктов, в котором нативный ген лактатдегидрогеназы или его часть были удалены. 16. Способ получения этанола, включающий культивирование микроорганизма согласно любому из предыдущих пунктов в подходящих условиях в присутствии С 3, С 5 или С 6 сахаров или состоящих из них олигомеров. 17. Способ согласно п.16, который осуществляют при температуре в пределах 40-70 С. 18. Способ согласно п.17, который осуществляют при температуре в пределах от 52 до 65 С. 19. Способ согласно любому из пп.16-18, согласно которому рН культуральной среды находится в пределах от 4 до 7,5. 20. Источник корма для животных, включающий микроорганизм согласно любому из пп.1-15.
МПК / Метки
Метки: термофильный, использовании, микроорганизм, выработки, способ, получения, модифицированный, этанола, повышенной
Код ссылки
<a href="https://eas.patents.su/28-17548-termofilnyjj-mikroorganizm-modificirovannyjj-dlya-povyshennojj-vyrabotki-etanola-i-sposob-polucheniya-etanola-pri-ego-ispolzovanii.html" rel="bookmark" title="База патентов Евразийского Союза">Термофильный микроорганизм, модифицированный для повышенной выработки этанола, и способ получения этанола при его использовании</a>
Способ стабильной непрерывной выработки этанола

Номер патента: 6106
Опубликовано: 25.08.2005
Авторы: Гадди Джеймз Л., Базу Рахул, Арора Динеш К., Уикстром Карл В., Ко Чинг-Ван, Филлипс Джон Рандалл, Клосен Эдгар К.
МПК: C12P 7/06
Метки: выработки, способ, стабильной, непрерывной, этанола
Формула / Реферат:
1. Способ стабильной непрерывной выработки этанола путем анаэробной бактериальной ферментации газообразного субстрата, включающий культивирование в ферментационном устройстве анаэробных, ацетогенных бактерий, которые способны вырабатывать этанол, в жидкой питательной среде, в состав которой входит пантотенат кальция и которая имеет уровень pH менее 5; подачу в упомянутое устройство газообразного субстрата, в состав которого входит монооксид...
Термофильный микроорганизм, продуцирующий полиаспарагиновую кислоту или ее сополимер, и способы обработки скважин

Номер патента: 11228
Опубликовано: 27.02.2009
Авторы: Котлар Ханс Кристиан, Хёуген Ярле Андре
МПК: C12N 15/00
Метки: способы, полиаспарагиновую, микроорганизм, скважин, продуцирующий, сополимер, кислоту, термофильный, обработки
Формула / Реферат:
1. Термофильный микроорганизм, который создан путем генной инженерии для того, чтобы быть способным продуцировать полиаспарагиновую кислоту или ее сополимер. 2. Микроорганизм по п.1, отличающийся тем, что представляет собой архебактерию. 3. Микроорганизм по п.1, где указанный микроорганизм также является галофильным и анаэробным. 4. Микроорганизм по п.1, где полиаспарагиновая кислота или ее сополимер секретируются микроорганизмом. 5. Пористая...
Способ получения этанола

Номер патента: 876
Опубликовано: 26.06.2000
Авторы: Ежков Александр Викторович, Арсеньев Дмитрий Викторович, Глюков Александр Анатольевич, Кузмичев Дмитрий Андреевич, Красницкий Владислав Михайлович
МПК: B01D 3/00
Метки: способ, получения, этанола
Формула / Реферат:
Способ получения этанола, предусматривающий выделение из бражки легколетучих компонентов и паров этанола с последующей их конденсацией и проведением процесса в герметичной емкости, снабженной вакуум-насосом и сборниками конденсатов, отличающийся тем, что бражку в процессе брожения нагревают до оптимальной температуры для используемой расы дрожжей и одновременно осуществляют отгонку этанола до окончания процесса спиртообразования. ...
Микроорганизм для детоксикации фумонизинов, а также его применение, способ детоксикации фумонизинов и кормовая добавка, содержащая этот микроорганизм

Номер патента: 13566
Опубликовано: 30.06.2010
Авторы: Тойбель Мартин, Фекиру Элизавет, Биндер Эва-Мария, Шатцмаир Герд
МПК: A23L 1/015, A23K 1/00
Метки: фумонизинов, также, способ, детоксикации, добавка, применение, содержащая, микроорганизм, кормовая
Формула / Реферат:
1. Микроорганизм для детоксикации фумонизинов и производных фумонизина, выбранный из группы, включающей штаммы Sphingopyxis sp. (DSM 16254 и DSM 16257), принадлежащие к таксону Sphingomonadaceae; штамм Ochrobactrum sp. (DSM 16255), принадлежащий к таксону Rhizobiales; штамм Leucobacter sp. (DSM 16256), принадлежащий к таксону Microbacteriaceae; штамм Agrobacterium sp. (DSM 16253), принадлежащий к таксону Rhizobiales; штамм Alcaligenes sp. (DSM...
Способ получения 1-(3,4-диметоксифенил) этанола

Номер патента: 2328
Опубликовано: 25.04.2002
Авторы: Берток Бэла, Арваи Геза, Салай Эржебет, Куруцне Рибаи Жужанна, Секей Иштван, Аради Матьяш
МПК: C07C 41/26
Метки: этанола, способ, получения, 1-(3,4-диметоксифенил
Формула / Реферат:
1. Способ получения 1-(3,4-диметоксифенил)этанола путем восстановления 3,4-диметоксиацетофенона формулы II, отличающийся тем, что карбонильную группу 3,4-диметоксиацетофенона восстанавливают водородом в присутствии протонного растворителя, предпочтительней в водной среде в условиях каталитического гидрирования. 2. Способ по п.1, отличающийся тем, что восстановление проводят в присутствии катализатора никеля Ренея. 3. Способ по пп.1-2,...
Предыдущий патент: Система и способ получения хлорбензола (варианты)
Следующий патент: Стабильные жидкие композиции антител против вируса бешенства
Случайный патент: Антисептическая композиция для дезинфекции кожных покровов, способ ее приготовления и полупродукт для ее приготовления