Способ получения (3s,3s’) 4,4′-дисульфандиилбис(3-аминобутан-1-сульфокислоты)
Номер патента: 21612
Опубликовано: 30.07.2015
Авторы: Кртиньи Йохан, Пти Мари-Ноэль, Мадек Джонатан, Шнайдер Жан-Мари, Балавуан Фабрис, Кокрель Жерар, Кувра Никола









Формула / Реферат
1. Способ получения (3S,3S') 4,4'-дисульфандиилбис(3-аминобутан-1-сульфокислоты) из (S) этил 2-(бензилоксикарбониламино) 4-(неопентилоксисульфонил)бутаноата А, включающий стадии, на которых:
(a) сложный этиловый эфир А восстанавливают с получением (S) неопентил 3-(бензилоксикарбониламино) 4-гидроксибутан-1-сульфоната В;
(b) спирт В вводят в реакцию с метансульфоновым ангидридом или метансульфонилхлоридом в присутствии основания с получением (S) неопентил 3-(бензилоксикарбониламино) 4-(метилсульфонилокси)бутан-1-сульфоната С;
(c) мезилированный спирт С вводят в реакцию с тиоацетатом калия с получением (S) 2-(бензилоксикарбониламино) 4-(неопентилоксисульфонил)бутил тиоацетата D;
(d) D димеризуют с получением (3S,3S') неопентил 4,4'-дисульфандиилбис(3-(бензилоксикарбониламино)бутан-1-сульфоната) Е;
(e) с Е снимают защитные группы сложного сульфонового эфира и амина с получением (3S,3S') 4,4'-дисульфандиилбис(3-аминобутан-1-сульфокислоты).
2. Способ по п.1, в котором стадию (а) осуществляют путем взаимодействия А с парой восстановитель - растворитель, выбранной из NaBH4/LiCl - смесь THF и этанола, и LiBH4 - THF при температуре от примерно 0 до примерно 25°C.
3. Способ по п.1, в котором стадию (а) осуществляют путем взаимодействия А с LiBH4 - THF при температуре от примерно 20 до примерно 25°C.
4. Способ по любому из пп.1-3, в котором стадию (b) осуществляют в присутствии триэтиламина в качестве основания в растворителе, выбранном из хлороформа и смеси МТВЕ и толуола, при температуре от примерно -10 до примерно 10°C.
5. Способ по п.1, в котором стадию (b) осуществляют путем взаимодействия В с метансульфонилхлоридом в присутствии триэтиламина в качестве основания в смеси МТВЕ и толуола с объемным отношением 3:2 при температуре от примерно 5 до примерно 10°C.
6. Способ по любому из пп.1-5, в котором стадию (с) осуществляют в растворителе, выбранном из этанола и ацетона.
7. Способ по любому из пп.1-5, в котором стадию (с) осуществляют в ацетоне при температуре от примерно 15 до примерно 25°C.
8. Способ по любому из пп.1-7, в котором стадию (d) осуществляют путем приведения D в контакт с гидроксидом натрия в этаноле и взаимодействия полученной смеси с йодом в этаноле при температуре от примерно 15 до примерно 25°C.
9. Способ по любому из пп.1-8, в котором стадию (е) осуществляют путем перемешивания Е в смеси TFA и анизола.
10. Способ по п.9, в котором стадию (е) осуществляют путем перемешивания Е в нагреваемой с обратным холодильником смеси TFA и анизола с объемным отношением 5:1.
11. Способ по любому из пп.1-10, в котором очистку конечного продукта осуществляют путем перекристаллизации в воде.
12. Способ по любому из пп.1-11, в котором конечный продукт получают в виде одной из его гидратных форм, предпочтительно в виде его тригидратной формы.
13. Способ по любому из предшествующих пп.1-12, в котором А получают из L-гомоцистина способом, включающим стадии, на которых:
(а-1) L-гомоцистина вводят в реакцию с бензилхлорформиатом с получением (2S,2S') 4,4'-дисульфандиилбис(2-(бензилоксикарбониламино)бутановой кислоты) F;
(b-1) осуществляют реакцию этерификации между F и этанолом с получением (2S,2S') диэтил 4,4'-дисульфандиилбис(2-(бензилоксикарбониламино)бутаноата) G;
(с-1) осуществляют окислительное расщепление дисульфидной связи G с получением (S) этил 2-(бензилоксикарбониламино) 4-(хлорсульфонил)бутаноата Н и
(d-1) H вводят в реакцию с неопентиловым спиртом с получением (S) этил 2-(бензилоксикарбониламино) 4-(неопентилоксисульфонил)бутаноата А.
14. Способ по п.13, в котором стадию (а-1) осуществляют в THF в присутствии гидроксида натрия при температуре от примерно 15 до примерно 25°C.
15. Способ по любому из пп.13, 14, в котором стадию (b-1) осуществляют путем взаимодействия F с тионилхлоридом в чистом этаноле при температуре от примерно 45 до примерно 55°C.
16. Способ по любому из пп.13-15, в котором стадию (с-1) осуществляют путем взаимодействия G с хлором в этаноле при температуре от примерно 5 до примерно 10°C.
17. Способ по любому из пп.13-16, в котором стадию (d-1) осуществляют в присутствии триэтиламина в толуоле при температуре от примерно 15 до примерно 25°C.
18. Кристаллический тригидрат (3S,3S') 4,4'-дисульфандиилбис(3-аминобутан-1-сульфокислоты), проявляющий, по существу, следующие свойства в рентгеноструктурном анализе:

Текст
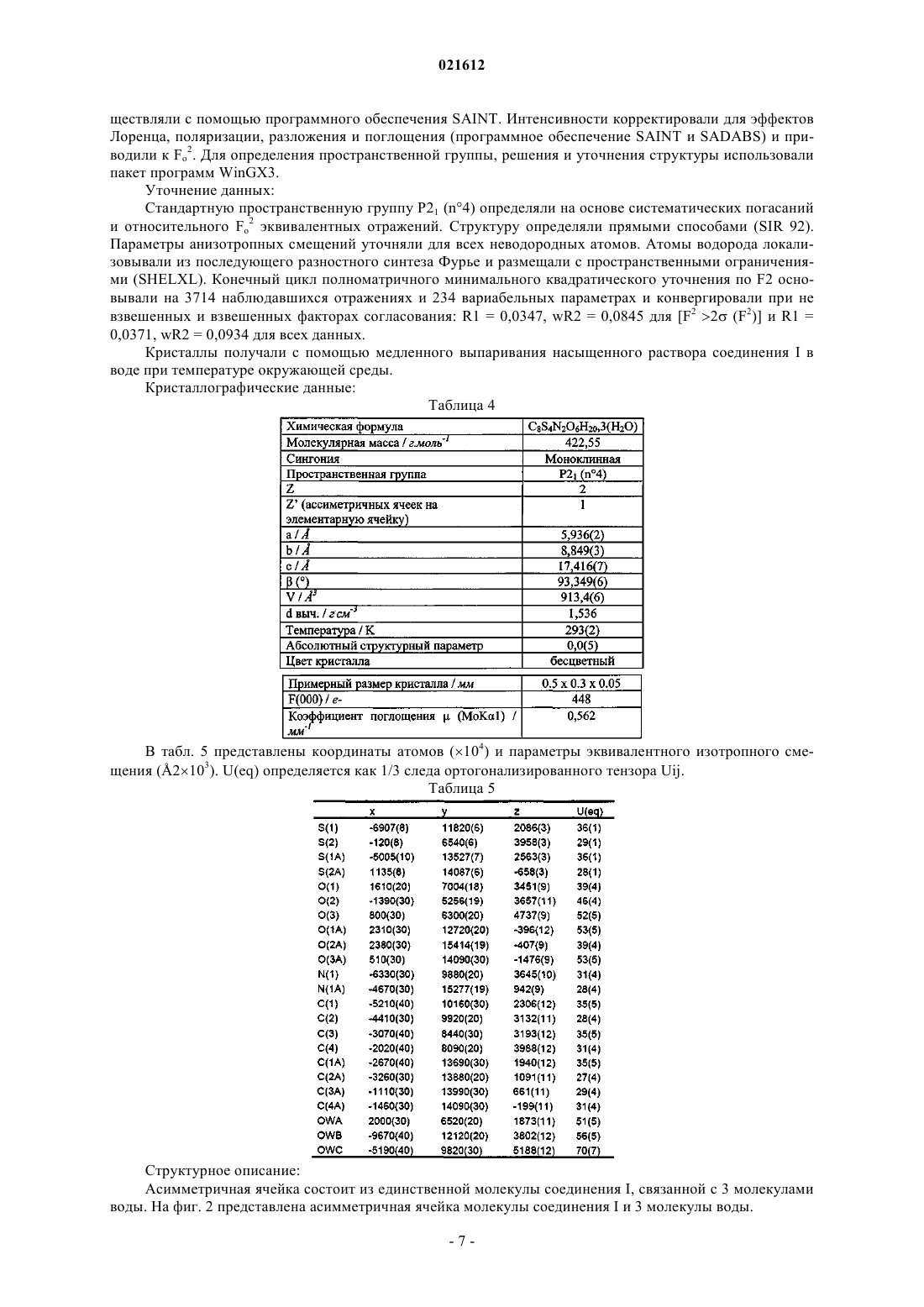
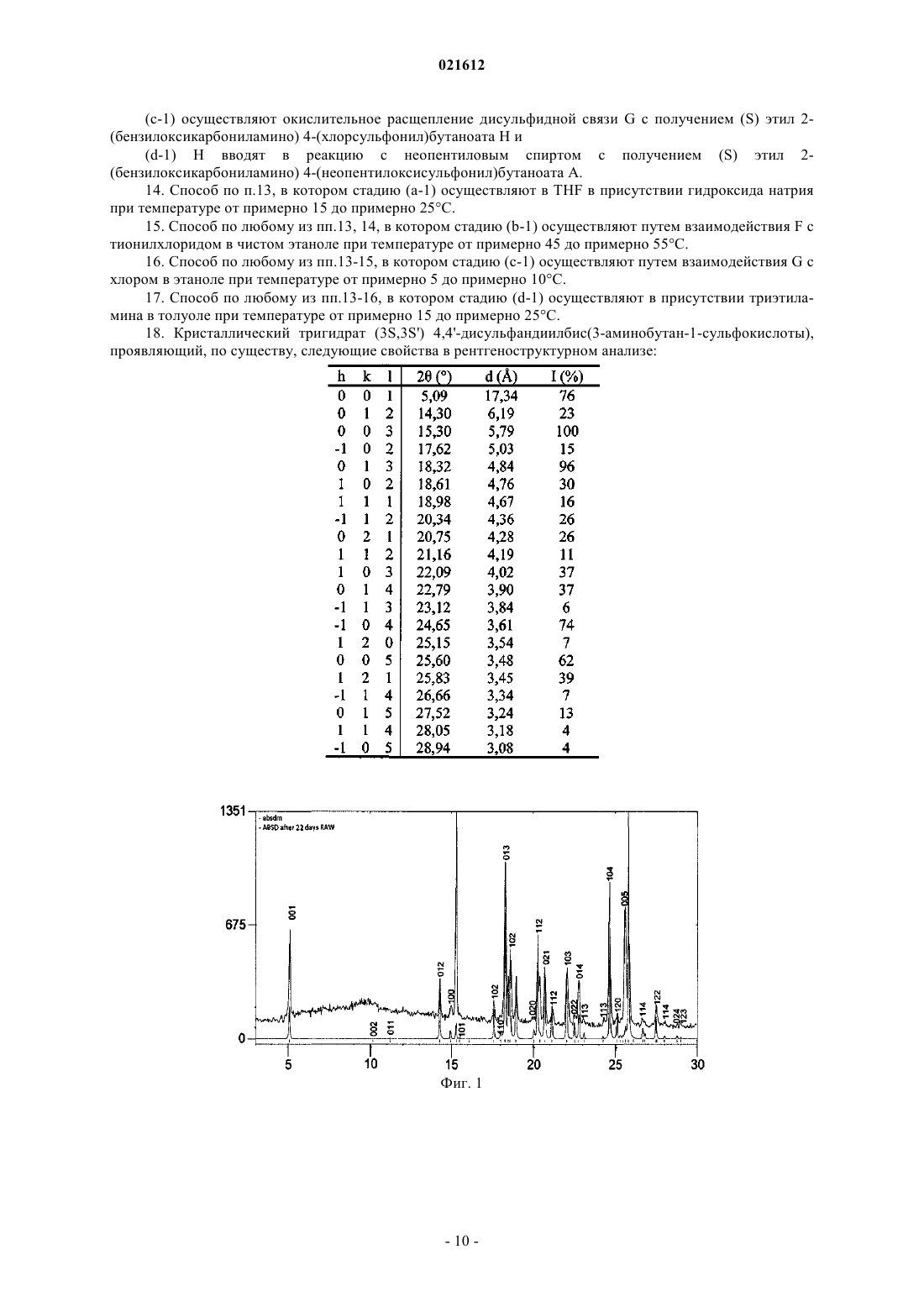
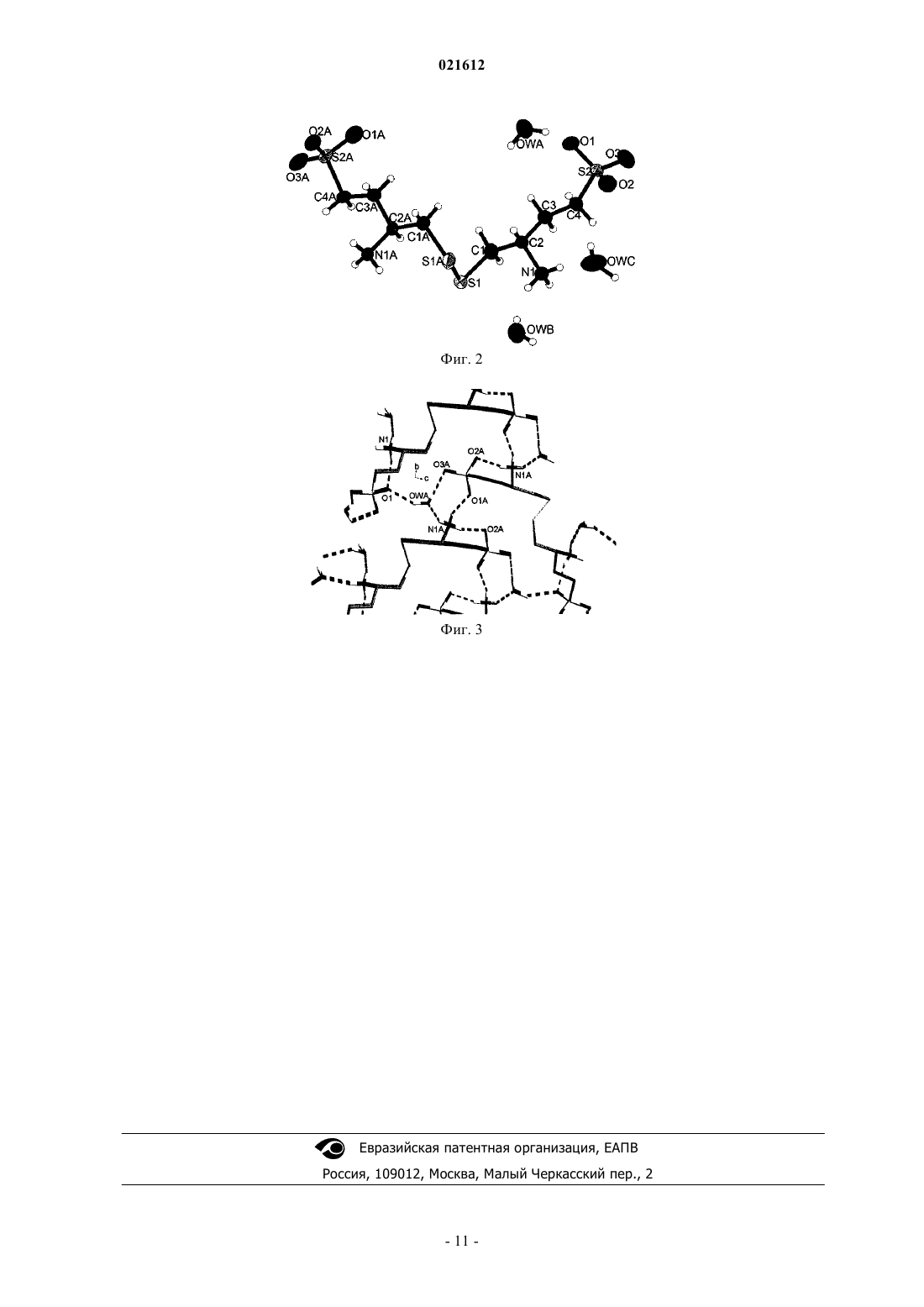
Настоящее изобретение относится к новому способу получения (3S,3S') 4,4'-дисульфандиилбис(3 аминобутан-1-сульфокислоты) в пять стадий из (S) этил 2-(бензилоксикарбониламино)-4(неопентилоксисульфонил)бутаноата А. Настоящее изобретение относится к новому способу получения (3S,3S') 4,4'-дисульфандиилбис(3 аминобутан-1-сульфокислоты) в пять стадий из (S)-этил 2-(бензилоксикарбониламино)-4(неопентилоксисульфонил)бутаноата A. (3S,3S') 4,4'-дисульфандиилбис(3-аминобутан-1-сульфокислота) именуется в настоящем изобретении "Соединение I". Соединение I является димером селективного ингибитора аминопептидазы А (АРА) 3-амино 4 меркаптобутансульфокислоты (также именуемой ЕС 33 в предшествующих документах), полученным путем образования дисульфидной связи между концевыми тиольными группами двух молекул 3-амино 4-меркаптобутансульфокислоты. Димеризация позволяет молекуле легче преодолевать гематоэнцефалический барьер в качестве пролекарства. Доказано, что соединение I (также именуемое в предшествующих документах RB150) является эффективным антигипертензивным агентом, как описано Bodineau etal., Hypertension 2008 51, 1318-1325. Соединение I и его использование в качестве антигипертензивного агента были описаны в патентной заявке WO 2004/007441. Пример способа синтеза соединения I, приведенный в данном документе,дает возможность получать целевое соединение в 6 стадий из L-гомосерина. Технические условия, в частности число эквивалентов, растворители и/или технологии очистки, применяемые в данном способе, не позволяют эффективно и легко перейти к его использованию в промышленном масштабе. В органическом синтезе постоянной целью является создание способов синтеза, которые могут быть использованы в промышленных условиях. Для соответствия требованиям, предъявляемым к промышленным процессам, необходимо оптимизировать различные параметры синтеза. Во-первых, растворитель должен быть как можно менее летучим, чтобы его можно было легко извлечь. Так, например,предпочтительно избегать использования хлорированных летучих растворителей, например дихлорметана, хлороформа и/или четыреххлористого углерода. Кроме того, число эквивалентов необходимых реагентов предпочтительно является ограниченным, применяемые температуры предпочтительно находятся в легкодоступном диапазоне, и предпочтение должно отдаваться легко осуществимым стадиям очистки. Наконец, реакционные смеси и выделенный продукт предпочтительно должны являться термически устойчивыми. Для получения лекарственных препаратов, предназначенных для человека или животных, были разработаны актуальные принципы добросовестного производства (с-GMP). Принципы GMP требуют качественного подхода к производству, что позволяет компаниям минимизировать или вовсе исключить случаи контаминации, путаницы и ошибок. Принципы GMP касаются вопросов, включающих ведение документооборота, квалификацию персонала, санитарию, чистоту, проверки оборудования, валидацию процесса и рассмотрение жалоб. Насколько известно заявителю, до настоящего момента не был описан ни один промышленно применимый процесс синтеза соединения I. В связи с этим, целью настоящего изобретения является разработка способа получения соединенияI, который может быть легко и эффективно применен в промышленном масштабе, если проводить сравнение со способом из известного уровня техники, в котором используются токсичные растворители, такие как диметилформамид, и колоночная хроматография. Более того, поскольку для лечения человека, как правило, необходима высокочистая форма, обычно более 99,5%, любого лекарственного препарата, способ, сочетающий возможность контроля образования изомеров и легкую конечную очистку продукта, является особенно предпочтительным. Способ Настоящее изобретение относится к новому способу получения соединения I, более конкретно в 5 стадий, из (S)-этил 2-(бензилоксикарбониламино)-4-(неопентилоксисульфонил)бутаноата А. Схема 1 иллюстрирует последовательные стадии, ведущие от А к соединению I. Если конкретно не указано иное, то во всем описании и во всей формуле настоящего изобретения используются следующие аббревиатуры и обозначения:THF = тетрагидрофуран; МТВЕ = метил-трет-бутиловый эфир; ВЭЖХ = высокоэффективная жидкостная хроматография; ее = энантиомерный избыток. Каждую реакцию, описанную в настоящем документе, можно осуществлять в твердой фазе или в жидкой фазе. Жидкофазные реакции предпочтительно можно осуществлять в растворителе, выбранном из органических или водных растворителей, например THF, этанола, хлороформа, МТВЕ, толуола, ацетона, TFA, и/или анизола. Первый аспект настоящего изобретения относится к общему способу получения соединения I из А,включающему следующие стадии:(a) восстановления сложного этилового эфира А для получения (S)-неопентил 3(бензилоксикарбониламино)-4-гидроксибутан 1-сульфоната В;(b) взаимодействия спирта В с метансульфоновым ангидридом или метансульфохлоридом в присутствии основания для получения(c) взаимодействия мезилированного спирта С с тиоацетатом калия для получения (S) 2(бензилоксикарбониламино) 4-(неопентилоксисульфонил)бутил тиоацетата D;(e) снятия защитных групп сложного сульфонового эфира и амина с Е для получения (3S,3S') 4,4'дисульфандиилбис(3-аминобутан-1-сульфокислоты) - соединения I. Вышеописанный способ именуется в настоящем описании "общим способом". Предпочтительно стадию (а) можно осуществлять посредством взаимодействия А с парой восстановитель-растворитель, выбранной из NaBH4/LiCl - смесь THF и этанола, предпочтительно с объемным соотношением 1:1, и LiBH4 - THF, более предпочтительно LiBH4 - THF. Реакцию можно осуществлять при температуре от примерно 0C до примерно 25C, предпочтительно от примерно 20C до примерно 25C. Более предпочтительно, стадию (а) можно осуществлять путем взаимодействия А с LiBH4 - THF при температуре от примерно 20 до примерно 25C. Использование растворимого и устойчивого в THF LiBH4 является бесспорным улучшением с точки зрения безопасности; в частности, это позволяет использовать в качестве растворителя чистый THF и,таким образом, избегать высвобождения газообразного водорода из-за разложения борогидрида натрия в этаноле. Предпочтительно стадию (b) можно осуществлять в присутствии триэтиламина. Реакцию можно осуществлять в растворителе, выбранном из хлороформа и смеси МТВЕ и толуола, предпочтительно смеси МТВЕ и толуола, предпочтительно с объемным соотношением 3:2. Реакцию можно осуществлять при температуре от примерно -10 до примерно 10C, предпочтительно от примерно 5 до примерно 10C. Переход к синтезу промышленного масштаба требует, чтобы летучие растворители предпочтительно были заменены менее летучими и/или легче извлекаемыми растворителями. Более предпочтительные условия для стадии (b) в данном изобретении включают замену хлороформа менее летучим и/или легче извлекаемым растворителем, таким как смесь МТВЕ и толуола с объемным отношением 3:2. Более предпочтительно стадию (b) можно осуществлять в присутствии триэтиламина в смеси МТВЕ и толуола с объемным отношением 3:2, при температуре от примерно 5 до примерно 10C. Предпочтительно стадию (с) можно осуществлять в растворителе, выбранном из этанола и ацетона,предпочтительно в ацетоне. Реакцию можно осуществлять при температуре от примерно 15 до примерно 25C. Более предпочтительно стадию (с) можно осуществлять в ацетоне при температуре от примерно 15 до примерно 25C. Предпочтительно стадию (d) можно осуществлять сначала путем контактирования D с гидроксидом натрия. Полученная смесь затем может реагировать с йодом. В качестве растворителя может использоваться этанол. Реакцию можно осуществлять при температуре от примерно 15 до примерно 25C. Предпочтительно стадию (е) можно осуществлять посредством перемешивания Е в смеси TFA и анизола. Более предпочтительно стадию (е) можно осуществлять посредством перемешивания Е в нагреваемой с обратным холодильником смеси из TFA и анизола, предпочтительно с объемным отношением 5:1. Наиболее предпочтительным вариантом осуществления настоящего изобретения является описанный выше общий способ, в котором стадию (а) осуществляют путем взаимодействия А с LiBH4 - THF при температуре от примерно 20 до примерно 25C; стадию (b) осуществляют в присутствии триэтиламина в смеси МТВЕ и толуола с объемным отношением 3:2 при температуре от примерно 5 до примерно 10C; стадию (с) осуществляют в ацетоне при температуре от примерно 15 до примерно 25C; стадию (d) осуществляют сначала путем контактирования D с гидроксидом натрия в этаноле при температуре от примерно 15 до примерно 25C, а затем путем взаимодействия полученной смеси с йодом в этаноле при температуре от примерно 15 до примерно 25C и стадию (е) осуществляют путем перемешивания Е в нагреваемой с обратным холодильником смесиTFA и анизола с объемным отношением 5:1. Данный способ включает стадии (а)-(е), оптимизированные для промышленного применения. В частности, стадии (b)-(е) даже соответствуют принципам c-GMP. Переход к синтезу промышленного масштаба требует оптимизации ряда параметров. В частности,предпочтительно избегать высокоэнтальпийных реакций. Для продуктов предпочтительна высокая степень чистоты. Выделяемые продукты предпочтительно должны быть термически устойчивыми. В табл. 1 приводится энтальпия реакций, чистота (определенная с помощью ВЭЖХ и выраженная в молярной процентной концентрации) и устойчивость продуктов на каждой стадии для данного предпочтительного способа. Таблица 1 Переход к синтезу промышленного масштаба требует, чтобы предпочтение отдавалось легким в осуществлении стадиям очистки, особенно это касается последней стадии очистки. Предпочтительный вариант осуществления данного изобретения относится к способу синтеза соединения I, как описано выше, в котором очистку соединения I осуществляют путем перекристаллизации в воде. Переход к синтезу промышленного масштаба требует, чтобы предпочтение отдавалось наиболее устойчивым формам соединений, особенно наиболее устойчивой форме конечного продукта. Исследования, выполненные для соединения I, показали, что гидратные формы, особенно тригидратная форма, являются более устойчивыми, чем чистая форма этого соединения. Тригидратная форма соединения I (3H2O) является наиболее устойчивой в условиях окружающей среды. Любая смесь гидратов соединения I в течение нескольких дней в условиях окружающей среды превращается в тригидратную форму. Под условиями окружающей среды в настоящем документе понимается температура между 15 и 25C при атмосферном давлении и относительной влажности выше 50%. Предпочтительный вариант осуществления данного изобретения относится к способу синтеза соединения I, как описано ранее, в котором соединение I получают в гидратной форме, предпочтительно в тригидратной форме. Другим аспектом настоящего изобретения, таким образом, является кристаллическая тригидратная форма соединения I. В частности, кристаллографическая структура тригидрата соединения I подробно описана в примере 2. Синтез исходного реагента для описанного выше общего способа, (S) этил -2(бензилоксикарбониламино) 4-(неопентилоксисульфонил)бутаноата А, уже описывался в патентной заявке WO 2004/007441, в качестве промежуточного продукта в ходе синтеза 4,4'-тиобис (3-аминобутан-1 неопентилсульфонат) бис-трифторацетата. Его выделение при необходимости может быть легко выполнено специалистом в данной области техники. Другим аспектом настоящего изобретения является описанный выше общий способ, в котором синтез соединения А из L-гомоцистина включает следующие стадии:(а-1) взаимодействия L-гомоцистина с бензилхлорформиатом для получения (2S,2S') 4,4'дисульфандиилбис(2-(бензилоксикарбониламино)бутановой кислоты) F;(b-1) осуществления реакции этерификации между F и этанолом для получения (2S,2S')-диэтил 4,4'дисульфандиилбис(2-(бензилоксикарбониламино)бутаноата) G;(с-1) окислительного расщепления дисульфидной связи G для получения (S) этил 2-3 021612(d-1) взаимодействия сульфонилхлорида Н с неопентиловым спиртом для получения (S) этил 2(бензилоксикарбониламино) 4-(неопентилоксисульфонил)бутаноата А. Синтез А из L-гомоцистина проиллюстрирован на схеме 2. Схема 2 При том же подходе, что и при синтезе соединения I из А, способ синтеза А можно оптимизировать,чтобы добиться максимально возможного соответствия промышленным требованиям. Предпочтительно стадию (а-1) можно осуществлять в присутствии гидроксида натрия. Растворителем может быть THF. Реакцию можно осуществлять при температуре от примерно 5 до примерно 25C,температуру в ходе добавления реагентов предпочтительно можно поддерживать между примерно 5 и примерно 10C. Легкость анализа является важным критерием того, подходит ли синтез для промышленности. Присутствие защитных аминогрупп Cbz в соединении F может сделать его более подходящим для анализа,особенно для ВЭЖХ-анализа. Предпочтительно стадию (b-1) можно осуществлять путем взаимодействия F с тионилхлоридом. Растворителем может быть чистый этанол. Реакцию можно осуществлять при температуре от примерно 45 до примерно 55C. Предпочтительно стадию (с-1) можно осуществлять путем взаимодействия G с хлором. Растворителем может быть этанол. Реакцию можно осуществлять при температуре от примерно 5 до примерно 10C. Предпочтительно стадию (d-1) можно осуществлять в присутствии триэтиламина. Растворителем может быть толуол. Реакцию можно осуществлять при температуре от примерно 15 до примерно 25C. Краткое описание чертежей Фиг. 1: Сравниваются порошковые рентгеновские дифрактограммы (XRPD) тригидрата соединения I: рассчитанная для монокристаллической структуры (нижний спектр) и экспериментальная (верхний спектр). Фиг. 2:ORTEP (Oak Ridge Thermal Ellipsoid Plot)-изображение тригидрата соединения I. Фиг. 3: Проекция вдоль оси тригидрата соединения I. Н-связи показаны пунктирными линиями. Примеры Пример 1. Синтез соединения I из (S) этил 2-(бензилоксикарбониламино) 4(неопентилоксисульфонил)бутаноата A. Стадия (а): (S) неопентил 3-(бензилоксикарбониламино) 4-гидроксибутан 1-сульфонат В(S) этил 2-(бензилоксикарбониламино)-4-(неопентилоксисульфонил)бутаноат А (41,55 г, 100,0 ммоль, 1,0 экв.) добавляют по каплям в 2 М раствор LiBH4 в THF (50 мл, 44,8 г, 100,0 ммоль, 1,0 экв.). Добавление осуществляют при комнатной температуре в течение 3 ч. После добавления смесь перемешивают при комнатной температуре до завершения конверсии (А 1%). Добавление толуола с последующими гидролизом с HCl, промывками органического слоя NaHCO3 и водой, и концентрированием под вакуумом, приводят к образованию с количественным выходом (ее = 98%) желаемого продукта в виде светло-желтого масла, которое медленно кристаллизуется при комнатной температуре за 4 или 5 дней. Так как с помощью ДСК-анализа обнаружено, что В имеет очень низкую температуру плавления, то его не удалось выделить в виде твердого вещества путем простой кристаллизации. Было решено оставить его в растворе и использовать без дальнейшей очистки на следующей стадии. Стадия (b): (S) неопентил 3-(бензилоксикарбониламино) 4-(метилсульфонилокси)бутан 1-сульфонат С Раствор В (57,64 г, 154,34 ммоль, 1,0 экв.) в толуоле (115 мл, 2,0 об.) разбавляют МТВЕ (173 мл, 3,0 об.) при комнатной температуре. Затем при комнатной температуре добавляют мезилхлорид (17,9 мл,26,5 г, 231,50 ммоль, 1,5 экв.), и однородную смесь охлаждают до 10C. Добавление триэтиламина (43,0 мл, 31,2 г, 308,67 ммоль, 2,0 экв.) осуществляют при Т 20C. После добавления смесь перемешивают при 10C до завершения конверсии (В 1%). После гидролиза с разбавленной HCl органический слой промывают NaHCO3, водой и насыщенным солевым раствором с последующим частичным концентрированием при пониженном давлении. Соответствующий мезилат далее кристаллизуют путем добавления гептана(5,0 об.) при 40C. После охлаждения, фильтрации и высушивания целевой продукт выделяют в виде беловатого твердого вещества с выходом 92,5% и очень высокой химической чистотой (98%). Стадия (с): (S) 2-(бензилоксикарбониламино) 4-(неопентилоксисульфонил)бутил тиоацетат D Раствор мезилата С (81,3 г, 180,05 ммоль, 1,0 экв.) в ацетоне (203 мл, 2,5 об.) добавляют по каплям к суспензии тиоацетата калия (41,1 г, 360,1 ммоль, 2,0 экв.) в ацетоне (203 мл, 2,5 об.) при комнатной температуре в течение 2 ч. Реакционную смесь перемешивают при комнатной температуре до завершения конверсии (С 1%). После фильтрации солей и добавления толуола (4,0 об.) ацетон удаляют с помощью дистилляции при пониженном давлении и 25C. Далее раствор очищают с активированным углем и концентрируют до 2,0 объемов. Медленное добавление гептана (5,0 об.) при комнатной температуре с последующим охлаждением при 0C, фильтрацией и высушиванием при 45C, дают целевой продукт в виде беловатого твердого вещества с выходом 78,2% и очень высокой химической чистотой (98%). Стадия (d): (3S,3S') неопентил 4,4'-дисульфандиилбис(3-(бензилоксикарбониламино)бутан 1 сульфонат) Е Раствор D (59,16 г, 137,1 ммоль, 1,0 экв.), суспендированный в этаноле (203 мл, 2,5 об.), охлаждают до 0C. Далее 20% гидроксида натрия (25,1 мл, 150,8 ммоль, 1,1 экв.), разбавленного водой (16,9 мл,0,285 об.), добавляют по каплям к суспензии, поддерживая температуру ниже 10C. Реакционную смесь нагревают до комнатной температуры и перемешивают до завершения конверсии (D1%). Промежуточный тиол взаимодействует при комнатной температуре с раствором йода (20,9 г, 82,3 ммоль, 0,6 экв.) в этаноле (118 мл, 2,0 об.). Реакция завершается в конце добавления окислителя. После добавления водного раствора (118 мл, 2,0 об.) Na2S2O5 (13,0 г, 68,5 ммоль, 0,5 экв.) для уменьшения избытка остаточного йода этанол удаляют с помощью дистилляции при пониженном давлении и 40C. Добавление воды (3,0 об.) при комнатной температуре с последующим охлаждением при 0C, фильтрацией и высушиванием при 45-50C, дают целевой димер в виде белого твердого вещества с выходом 98,3% и очень высокой химической чистотой (97,0%). Количество иодид-ионов, образующихся при восстановлении йода, проверяется в образце потенциометрическим методом.E=E0(Ag+/Ag(s+0,06 log (KSI/[I-]) Анализ: [I-] понижается и Е возрастает Предел обнаружения=1 мг Далее выполняются четыре промывки водой до прекращения обнаружения иодид-ионов. Результаты представлены в табл. 2. Раствор Е (44,0 г, 56,6 ммоль, 1,0 экв.) в TFA (220 мл, 5,0 об.) и анизоле (44 мл, 1,0 об.) нагревают с обратным холодильником (75C) и реакционную смесь перемешивают в этих условиях до завершения конверсии (Е 1%). TFA удаляют с помощью дистилляции при пониженном давлении и 50C. Медленное добавление МТВЕ (5,0 об.) при комнатной температуре приводит к осаждению целевого продукта. После растирания в порошок, фильтрации и промывки с МТВЕ (1,0 об.) твердое сырое вещество суспендируют в метаноле (220 мл, 5,0 об.). Новое растирание в порошок, фильтрация и промывка с МТВЕ (1,0 об.), с последующей сушкой при пониженном давлении приводят к образованию соединения I в виде белого твердого вещества с выходом 92,5% . ЯМР: 1 Н (растворитель D2O, 400 МГц, м.д.): 4,70 (s, 6 Н, Н 5); 3,77 (m, 2 Н, Н 2); 3,14 (dd, 2H, Н,); 2,98(dd, 4H, Н 4); 2,86 (dd, 2H, H1); 2,13 (m, 4H, Н 3). 13 С (растворитель D2O, 100 МГц, м.д.): 49,4 (2 С, С 2); 46,6 (2 С, С 4); 38,3 (2 С, d); 26,9 (2 С, С 3). Пример 2. Кристаллографические данные тригидрата соединения I Соединение I, полученное в примере 1, хранили в течение 22 дней в условиях окружающей среды для получения чистой тригидратной формы. Сбор данных Кристаллическую структуру тригидрата соединения I [C8S4N2O6H2O, 3(Н 2 О)] определяли рентгеноструктурным анализом монокристалла (XRD). Выбранный кристалл наклеивали на стекловолокно и устанавливали на полный 3-кружный гониометр -дифрактометр Bruker SMART APEX с площадным детектором CCD. Записали три серии экспозиций (всего из 1800 кадров), соответствующие трем сканированиям (шаг по 0,3), для трех различных значений . На фиг. 1 представлены экспериментальная и расчетная XRD-дифрактограммы тригидрата соединения I. В табл. 3 представлена выборка расчетных рефлексов из порошковой ячейки для структуры тригидрата соединения I и соответствующие положения экспериментальных пиков, а также интенсивности. Величинам интенсивностей экспериментальных пиков не следует придавать слишком большое значение,поскольку на них очень большое влияние оказывают эффекты предпочтительной ориентации в процессе кристаллизации. Таблица 3 Параметры ячейки и матрицу ориентации кристалла предварительно определяли с использованием программного обеспечения SMART. Интегрирование данных и общее уточнение параметров ячеек осу-6 021612 ществляли с помощью программного обеспечения SAINT. Интенсивности корректировали для эффектов Лоренца, поляризации, разложения и поглощения (программное обеспечение SAINT и SADABS) и приводили к Fo2. Для определения пространственной группы, решения и уточнения структуры использовали пакет программ WinGX3. Уточнение данных: Стандартную пространственную группу P21 (n4) определяли на основе систематических погасаний и относительного Fo2 эквивалентных отражений. Структуру определяли прямыми способами (SIR 92). Параметры анизотропных смещений уточняли для всех неводородных атомов. Атомы водорода локализовывали из последующего разностного синтеза Фурье и размещали с пространственными ограничениями (SHELXL). Конечный цикл полноматричного минимального квадратического уточнения по F2 основывали на 3714 наблюдавшихся отражениях и 234 вариабельных параметрах и конвергировали при не взвешенных и взвешенных факторах согласования: R1 = 0,0347, wR2 = 0,0845 для [F2 2 (F2)] и R1 = 0,0371, wR2 = 0,0934 для всех данных. Кристаллы получали с помощью медленного выпаривания насыщенного раствора соединения I в воде при температуре окружающей среды. Кристаллографические данные: Таблица 4 В табл. 5 представлены координаты атомов (104) и параметры эквивалентного изотропного смещения (2103). U(eq) определяется как 1/3 следа ортогонализированного тензора Uij. Таблица 5 Структурное описание: Асимметричная ячейка состоит из единственной молекулы соединения I, связанной с 3 молекулами воды. На фиг. 2 представлена асимметричная ячейка молекулы соединения I и 3 молекулы воды. Вдоль оси b соседние молекулы соединения I взаимодействуют посредством двух типов водородных связей, возникающих между атомом кислорода O1 А и атомом водорода H(N1A) (d1,94 ) и между атомом кислорода O1 и атомом водорода H(N1) (d1,99 ). Вдоль оси а две соседние молекулы соединения I взаимодействуют посредством водородной связи между атомом кислорода O2 А и атомом водородаH(N1A) (d1,94 ). Эти взаимодействия, ориентированные в a и b направлениях, приводят к слоям, параллельным (110). Кроме того, молекула воды (OWA) встраивается между этими молекулами и образует три различных водородных связи: первая связывает атом кислорода OWA с атомом водорода H(N1A)(d2,02 ), вторая связывает атом кислорода O3 А с атомом водорода H(OWA) (d1,94 ), и последняя связывает атом кислорода O1 с H(OWA) (d1,97 ). Срезы (110) имеют толщину d001 (17,5 ). Различные взаимодействия внутри этих срезов вдоль оси a представлены более подробно на фиг. 3. Два соседних слоя взаимодействуют вдоль оси с через водородные связи, образованные с двумя другими молекулами воды OWB и OWC, расположенными в плоскостях (002). Атом кислорода молекулы воды OWB образует водородные связи с атомом водорода Н (из N1) (d2,00 ) из первого среза, и связан со следующим срезом посредством водородной связи с атомом кислорода O3 и атомом водорода Н (из OWB)(d1,92 ). Атом кислорода молекулы воды OWC образует водородную связь с атомом водорода Н (изN1) (d1,86 ) из первого среза, и затем присоединяется к соседнему срезу посредством двух водородных связей через взаимодействия с двумя атомами кислорода из сульфонатного фрагмента S2. Атом кислорода O2 и атом кислорода O3 взаимодействуют с двумя атомами водорода молекулы воды OWC с длиной связей, соответственно, d2,30 и d2,03 . Пример 3. Синтез (S) этил 2-(бензилоксикарбониламино) 4-(неопентилоксисульфонил)бутаноата А из L-гомоцистина. Стадия (a-1): (2S,2S') 4,4'-дисульфандиилбис(2-(бензилоксикарбониламино)бутановая кислота) FL-гомоцистин (200,0 г, 745,5 ммоль, 1,0 экв.) суспендируют в THF (1000 мл, 5,0 об.) и охлаждают до 5-10C. После добавления 20% гидроксида натрия (521,7 мл, 626,2 г, 125,25 г при 100%, 3,13 ммоль, 4,2 экв.) добавляют бензилхлороформиат (220,3 мл, 267 г, 1565,5 ммоль, 2,1 экв.). Превращение завершают в течение ночи при комнатной температуре (L-гомоцистин 1%). Экстракции и промывки органического слоя водой приводят к желтому маслу, которое извлекают с количественным выходом, и которое медленно кристаллизуется в виде желтоватого твердого вещества при комнатной температуре. Стадия (b-1): (2S,2S') диэтил 4,4'-дисульфандиилбис(2-(бензилоксикарбониламино)бутаноат) GF (140,0 г, 260,89 ммоль, 1,0 экв.) суспендируют в чистом этаноле (700 мл, 5,0 об.) и нагревают до 50C. Добавление SOCl2 (41,6 мл, 68,3 г, 573,96 ммоль, 2,2 экв.) осуществляют при 50C во избежание его накопления. Первращение завершается (F1%) спустя 1 ч при 50C. Концентрирование сырой смеси с последующим растворением в этилацетате и промывками органического слоя приводят к образованию прозрачного раствора, который частично концентрируют. Медленное добавление 5,0 объемов гептана приводит к кристаллизации целевого продукта в виде белого твердого вещества. После фильтрации и высушивания выделяют сложный бис-эфир с выходом 92%. Стадия (с-1): (S) этил 2-(бензилоксикарбониламино)-4-(хлорсульфонил)бутаноат НG (100,0 г, 168,7 ммоль, 1,0 экв.) суспендируют в этаноле (500 мл, 5,0 об.) и охлаждают до 5C. Добавление Cl2 (83,7 г, 1,18 моль, 7,5 экв.) выполняют при Т 10C. Конверсия завершается (G1%), когда реакционная смесь становится совершенно однородной. Раствор сульфонилхлорида выливают в смесь водного раствора карбоната и толуола, поддерживая температуру ниже 20C. Промывки органического слоя, за которыми следует концентрирование при пониженном давлении, приводят к образованию целевого продукта в виде бесцветного масла с выходом 96,8%. Целевой продукт можно выделить в виде белого твердого вещества, если медленно добавлять 5,0 объемов гептана к концентрированному раствору (2,0 об.) продукта в толуоле. Тем не менее, чистота при этом существенно не улучшается, а выход резко понижается (75-80%). В результате, сульфонил хлорид Н выделяют в 1,0 объеме толуола и используют без дальнейшей очистки на следующей стадии. Стадия (d-1): (S) этил 2-(бензилоксикарбониламино) 4-(неопентилоксисульфонил)бутаноат А Неопентиловый спирт (29,1 г, 329,84 ммоль, 1,2 экв.) растворяют в толуоле (400 мл, 4,0 об.) и при комнатной температуре добавляют раствор Н (100,0 г, 274,87 ммоль, 1,0 экв.) в толуоле (100 мл, 1,0 об.). Однородную смесь затем охлаждают до 0C. При 0C добавляют триэтиламин (46,0 мл, 33,4 г, 329,84 ммоль, 1,2 экв.). После добавления смесь нагревают до комнатной температуры до завершения конверсии (Н 1%). После гидролиза с разбавленной HCl органический слой промывают NaHCO3, водой и насыщенным солевым раствором и концентрируют при пониженном давлении для получения целевого продукта в виде светло-желтого масла с выходом 94,4%. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения (3S,3S') 4,4'-дисульфандиилбис(3-аминобутан-1-сульфокислоты) из (S) этил 2(бензилоксикарбониламино) 4-(неопентилоксисульфонил)бутаноата А, включающий стадии, на которых:(a) сложный этиловый эфир А восстанавливают с получением (S) неопентил 3(бензилоксикарбониламино) 4-гидроксибутан-1-сульфоната В;(b) спирт В вводят в реакцию с метансульфоновым ангидридом или метансульфонилхлоридом в присутствии основания с получением (S) неопентил 3-(бензилоксикарбониламино) 4(метилсульфонилокси)бутан-1-сульфоната С;(e) с Е снимают защитные группы сложного сульфонового эфира и амина с получением (3S,3S') 4,4'-дисульфандиилбис(3-аминобутан-1-сульфокислоты). 2. Способ по п.1, в котором стадию (а) осуществляют путем взаимодействия А с парой восстановитель - растворитель, выбранной из NaBH4/LiCl - смесь THF и этанола, и LiBH4 - THF при температуре от примерно 0 до примерно 25C. 3. Способ по п.1, в котором стадию (а) осуществляют путем взаимодействия А с LiBH4 - THF при температуре от примерно 20 до примерно 25C. 4. Способ по любому из пп.1-3, в котором стадию (b) осуществляют в присутствии триэтиламина в качестве основания в растворителе, выбранном из хлороформа и смеси МТВЕ и толуола, при температуре от примерно -10 до примерно 10C. 5. Способ по п.1, в котором стадию (b) осуществляют путем взаимодействия В с метансульфонилхлоридом в присутствии триэтиламина в качестве основания в смеси МТВЕ и толуола с объемным отношением 3:2 при температуре от примерно 5 до примерно 10C. 6. Способ по любому из пп.1-5, в котором стадию (с) осуществляют в растворителе, выбранном из этанола и ацетона. 7. Способ по любому из пп.1-5, в котором стадию (с) осуществляют в ацетоне при температуре от примерно 15 до примерно 25C. 8. Способ по любому из пп.1-7, в котором стадию (d) осуществляют путем приведения D в контакт с гидроксидом натрия в этаноле и взаимодействия полученной смеси с йодом в этаноле при температуре от примерно 15 до примерно 25C. 9. Способ по любому из пп.1-8, в котором стадию (е) осуществляют путем перемешивания Е в смеси TFA и анизола. 10. Способ по п.9, в котором стадию (е) осуществляют путем перемешивания Е в нагреваемой с обратным холодильником смеси TFA и анизола с объемным отношением 5:1. 11. Способ по любому из пп.1-10, в котором очистку конечного продукта осуществляют путем перекристаллизации в воде. 12. Способ по любому из пп.1-11, в котором конечный продукт получают в виде одной из его гидратных форм, предпочтительно в виде его тригидратной формы. 13. Способ по любому из предшествующих пп.1-12, в котором А получают из L-гомоцистина способом, включающим стадии, на которых:(b-1) осуществляют реакцию этерификации между F и этанолом с получением (2S,2S') диэтил 4,4'дисульфандиилбис(2-(бензилоксикарбониламино)бутаноата) G;(с-1) осуществляют окислительное расщепление дисульфидной связи G с получением (S) этил 2(бензилоксикарбониламино) 4-(хлорсульфонил)бутаноата Н и(d-1) H вводят в реакцию с неопентиловым спиртом с получением (S) этил 2(бензилоксикарбониламино) 4-(неопентилоксисульфонил)бутаноата А. 14. Способ по п.13, в котором стадию (а-1) осуществляют в THF в присутствии гидроксида натрия при температуре от примерно 15 до примерно 25C. 15. Способ по любому из пп.13, 14, в котором стадию (b-1) осуществляют путем взаимодействия F с тионилхлоридом в чистом этаноле при температуре от примерно 45 до примерно 55C. 16. Способ по любому из пп.13-15, в котором стадию (с-1) осуществляют путем взаимодействия G с хлором в этаноле при температуре от примерно 5 до примерно 10C. 17. Способ по любому из пп.13-16, в котором стадию (d-1) осуществляют в присутствии триэтиламина в толуоле при температуре от примерно 15 до примерно 25C. 18. Кристаллический тригидрат (3S,3S') 4,4'-дисульфандиилбис(3-аминобутан-1-сульфокислоты),проявляющий, по существу, следующие свойства в рентгеноструктурном анализе:
МПК / Метки
МПК: C07C 309/14, C07C 303/02
Метки: 4,4'-дисульфандиилбис(3-аминобутан-1-сульфокислоты, 3s,3s, получения, способ
Код ссылки
<a href="https://eas.patents.su/12-21612-sposob-polucheniya-3s3s-44-disulfandiilbis3-aminobutan-1-sulfokisloty.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения (3s,3s’) 4,4′-дисульфандиилбис(3-аминобутан-1-сульфокислоты)</a>
Способ получения дихлорпропанола, способ получения эпихлоргидрина, способ получения эпоксидных смол и применение оборудования, обладающего коррозионной стойкостью, в способе получения дихлорпропанола

Номер патента: 14241
Опубликовано: 29.10.2010
Авторы: Краффт Филипп, Бальтазар Доминик, Сметс Валентин, Жильбо Патрик, Франк Кристиан
МПК: C07C 29/62, C07C 31/36, B01J 19/02...
Метки: коррозионной, стойкостью, способ, получения, применение, эпоксидных, дихлорпропанола, обладающего, оборудования, эпихлоргидрина, смол, способе
Формула / Реферат:
1.Способ получения дихлорпропанола, содержащий:(a) стадию, на которой глицерин или сложный эфир глицерина или их смесь вводят во взаимодействие с агентом хлорирования, содержащим хлороводород,(b) по меньшей мере одну другую стадию, осуществляемую на оборудовании, выполненном или имеющем покрытие из материалов, обладающих стойкостью по отношению к агенту хлорирования, в условиях осуществления этой стадии,причем другая стадия является стадией...
Способ получения дихлорпропанола, способ получения эпихлоргидрина и способ получения эпоксидных смол

Номер патента: 13681
Опубликовано: 30.06.2010
Авторы: Краффт Филипп, Жильбо Патрик
МПК: C07C 29/62, C07C 31/36, C07C 31/42...
Метки: получения, дихлорпропанола, смол, способ, эпихлоргидрина, эпоксидных
Формула / Реферат:
1. Способ получения дихлорпропанола, в котором вводят во взаимодействие глицерин, или сложный эфир глицерина, или их смесь, общее содержание металлов в которых, выраженное в расчете на элементы, выше или равно 0,1 мкг/кг и ниже или равно 1000 мг/кг, и агент хлорирования.2. Способ по п.1, в котором общее содержание металлов ниже или равно 500 мг/кг и который характеризуется по меньшей мере одним из следующих признаков:содержание железа в...
Цеолитный катализатор l-типа, способ его получения, способ получения ароматических углеводородов, способ получения бензина

Номер патента: 3559
Опубликовано: 26.06.2003
Авторы: Сугимото Митио, Фукунага Тецуя, Иннес Роберт А.
МПК: B01J 29/61, C07C 5/41, C10G 35/095...
Метки: способ, катализатор, углеводородов, получения, бензина, цеолитный, l-типа, ароматических
Формула / Реферат:
1. Цеолитный катализатор L-типа, который получают при нанесении на цеолит L-типа платинового компонента, одного или более галогеновых компонентов и одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, при этом наносимое количество одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, находится в интервале от 0,001 до 3 мас.% из расчета на общую массу катализатора, молярное отношение...
Катализатор для получения сложных эфиров,способ получения сложного эфира и способ получения сложного полиэфира с участием такого катализатора

Номер патента: 11171
Опубликовано: 27.02.2009
Авторы: Хэнратти Алан Джозеф, Партридж Мартин Грэхэм, Макинтош Кэлам Гарри
МПК: C08G 63/85, B01J 31/04
Метки: эфиров,способ, такого, катализатор, полиэфира, получения, способ, сложных, сложного, катализатора, участием, эфира
Формула / Реферат:
1. Катализатор для получения сложного эфира в реакции этерификации, состоящий из продукта взаимодействия: a) соединения титана, циркония или гафния; b) 2-оксикарбоновой кислоты; c) четвертичного аммониевого соединения, выбранного из группы, состоящей из гидроксида тетраэтиламмония и гидроксида тетраметиламмония, и d) соединения цинка. 2. Катализатор по п. 1, в котором соединением титана, циркония или гафния является алкоголят, имеющий формулу...
Способ получения полиолефина, применение смеси по меньшей мере двух агентов переноса цепи для получения полиолефина, полиэтилен низкой плотности, способ получения кабеля и кабель

Номер патента: 19226
Опубликовано: 28.02.2014
Авторы: Смедберг Анника, Кампус Альфред, Шильд Герман, Хубер Маркус, Нильссон Ульф, Воигт Бьерн
МПК: C08L 23/06, C08F 10/02, C08F 2/38...
Метки: мере, двух, меньшей, переноса, полиэтилен, полиолефина, получения, низкой, агентов, способ, кабеля, смеси, кабель, цепи, плотности, применение
Формула / Реферат:
1. Способ получения полиолефина, включающий полимеризацию по меньшей мере одного олефина в присутствии смеси по меньшей мере двух агентов переноса цепи, при этом смесь включаетполярный агент переноса цепи (полярный АПЦ) инеполярный агент переноса цепи (неполярный АПЦ), который представляет собой неароматический линейный, разветвленный или циклический углеводород, возможно содержащий гетероатом, такой как О, N, S, Si или Р, и не содержит полярной...
Предыдущий патент: Катализатор ионно-жидкостного состава для получения поли-альфа-олефиновых синтетических базовых смазочных масел
Следующий патент: Азабензимидазолы в качестве противовирусных средств в отношении респираторного синцитиального вируса
Случайный патент: Противоопухолевая терпеноидная фармацевтическая композиция с ангиогенезингибирующим действием