Способ получения гидрохинонов
Номер патента: 22721
Опубликовано: 29.02.2016









Формула / Реферат
1. Способ получения соединения гидрохинона формулы (I)

в которой R2, R3, R5, R6 независимо друг от друга означают Н, А, Сус, Hal, CN, -(CYY)n-OA, -(CYY)n-NYY, -O(CYY)n-OA, -O(CYY)n-NYY, -NH(CYY)n-OA или -NH(CYY)n-NYY;
Y означает Н, А или Hal;
А означает неразветвленный или разветвленный алкил, имеющий 1-10 атомов С, в котором 1-7 атомов Н могут быть замещены Hal и/или в котором одна или две соседние СН2 группы могут быть заменены независимо друг от друга -СН=СН- и/или -СºС- группой;
Сус означает циклоалкил, имеющий 3-7 атомов С, в котором 1-4 атома Н могут быть заменены независимо друг от друга A, Hal и/или OY;
Hal означает F, Cl, Br или I и
n равно 0, 1, 2, 3, 4, 5 или 6;
который включает стадии:
(а) нагревания до температуры кипячения с обратным холодильником соединения фенола формулы (II)

в которой R2, R3, R5 и R6 имеют значения, как определено выше, с 0,3-0,4 экв. гексаметилентетрамина в органической кислоте, с последующим добавлением гидролитической среды и нагреванием получившейся смеси до получения 4-гидроксибензальдегидного соединения формулы (III)

в которой R2, R3, R5 и R6 имеют значения, как определено выше, и
(b) реакции соединения формулы (III) с окислителем в растворителе в кислых условиях до получения соединения формулы (I).
2. Способ по п.1 получения соединения формулы (I), в которой
R2 означает Н или метил;
R3, R5 означают Н, метил, этил, пропил, изопропил, бутил или Hal;
R6 означает Н и
Hal означает F, Cl или Br.
3. Способ по п.2 для получения 2,3,5-триметилбензол-1,4-диола и/или 2,6-диэтилбензол-1,4-диола.
4. Способ по любому из пп.1-3, в котором на стадии (а) органической кислотой является ледяная уксусная кислота и/или гидролитическая среда представляет собой воду или серную кислоту.
5. Способ по любому из пп.1-4, в котором на стадии (а) нагревание осуществляют при температуре кипячения с обратным холодильником 110-130°С и/или на стадии (b) реакцию проводят при температуре между 15 и 50°С.
6. Способ по любому из пп.1-5, в котором на стадии (b) окислитель представляет собой пероксид водорода и/или растворителем является спирт.
7. Способ по любому из пп.1-6, в котором на стадии (b) кислотные условия обеспечивают серной кислотой в молярном соотношении к соединению формулы (III), составляющем 0,2.
8. Способ по любому из пп.1-7, в котором перед стадией (b) соединение формулы (III) очищают 0,1-10% гидроксидом натрия в молярном соотношении соединения формулы (III) к гидроксиду натрия от 0,2 до 5.
9. Способ по п.8, в котором перед стадией (b) соединение формулы (III) очищают 1-2% гидроксидом натрия в эквимолярном соотношении соединения формулы (III) к гидроксиду натрия.
10. Способ по любому из пп.1-9, в котором после стадии (b) соединение формулы (I) очищают 10-20% раствором дитионата натрия.
Текст
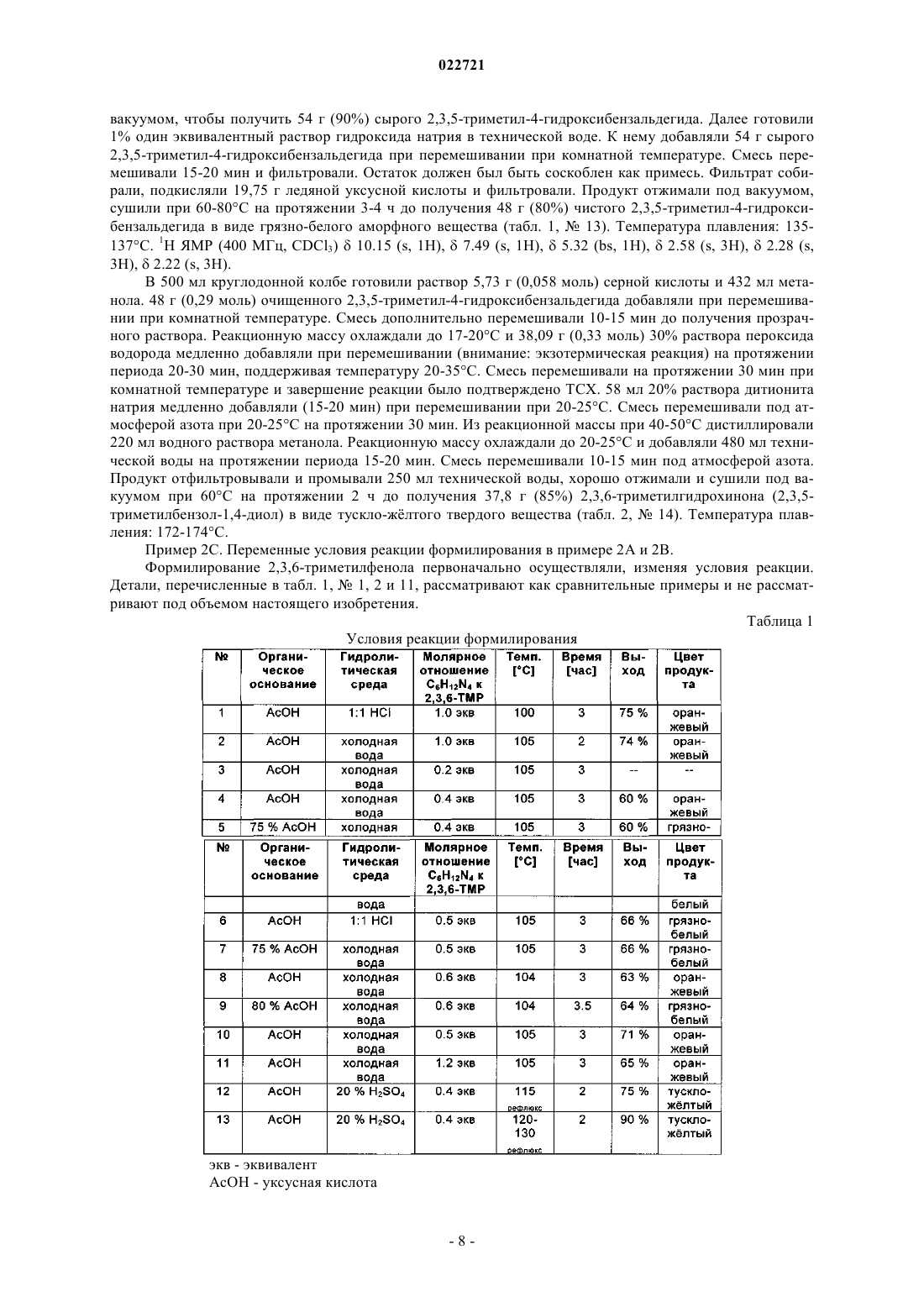
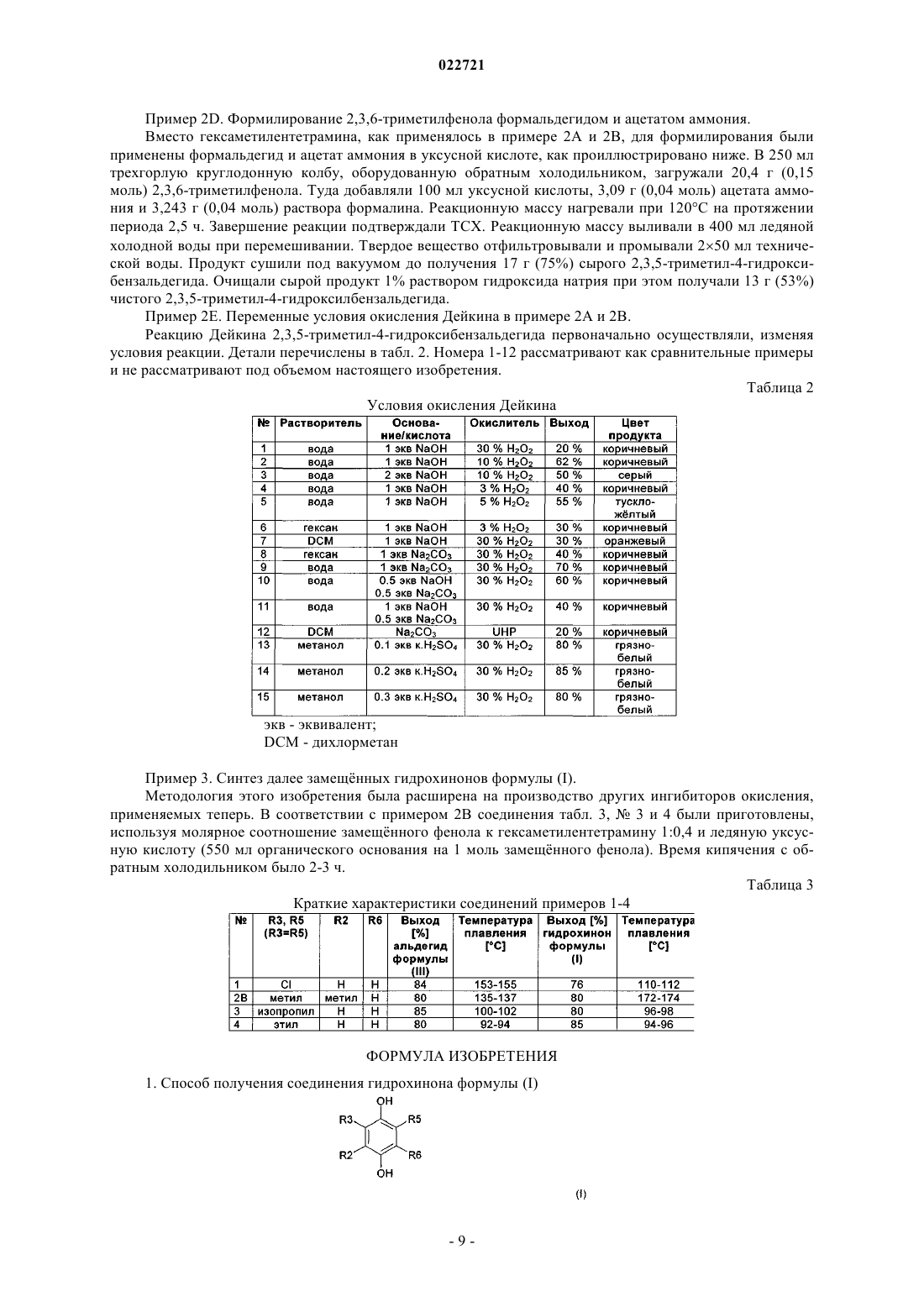
Изобретение относится к способу получения соединения гидрохинона формулы (I) в которой R2, R3, R5 и R6 имеют значения по п.1, со стадиями нагревания до температуры кипячения с обратным холодильником замещнного фенола с 0,3-0,4 экв. гексаметилентетрамина в органической кислоте, с последующим добавлением гидролитической среды и нагреванием получившейся смеси до выхода замещенного 4-гидроксибензальдегидного соединения и оксидирования полученного соединения в кислых условиях до соответствующего гидрохинона формулы (I).(71)(73) Заявитель и патентовладелец: МЕРК ПАТЕНТ ГМБХ (DE) Изобретение относится к способу получения соединения гидрохинона формулы (I) в которой R2, R3, R5 и R6 имеют значения по п.1, со стадиями нагревания до температуры кипячения с обратным холодильником замещнного фенола с 0,3-0,4 экв. гексаметилентетрамина в органической кислоте, с последующим добавлением гидролитической среды и нагреванием получившейся смеси до выхода замещенного 4-гидроксибензальдегидного соединения и оксидирования полученного соединения в кислых условиях до соответствующего гидрохинона формулы (I). Реакции окисления могут производить свободные радикалы, которые начинают цепные реакции,которые повреждают клетки. Ингибиторы окисления заканчивают эти цепные реакции, удаляя свободнорадикальные промежуточные соединения, и ингибируют другие реакции окисления, окисляясь при этом сами. Организмы содержат комплексную сеть антиокислительных метаболитов и ферментов, которые сотрудничают, чтобы предотвратить окислительное повреждение клеточных компонентов, таких как ДНК, белки и липиды. Поскольку окислительный стресс может быть важной частью многих человеческих болезней, в фармакологии интенсивно изучают применение ингибиторов окисления, особенно в качестве лечения инсульта и нейродегенеративных заболеваний. Ингибиторы окисления также широко применяются в качестве компонентов в пищевых добавках в надежде на поддержание здоровья и предотвращение болезней, таких как рак и ишемическая болезнь сердца. В дополнение к этому применению природных ингибиторов окисления в медицине, эти соединения имеют значительное промышленное применение, например консерванты в пище и косметических продуктах, также предотвращение ухудшения резины и бензин. Отдельным примером является 2-трет-бутилгидрохинон, который применяется в качестве ингибитора окисления в пищевых продуктах. Утверждается, что -токоферол является самым важным липидрастворимым ингибитором окисления и что он защищает клеточные мембраны от окисления, реагируя с липидными радикалами, полученными в цепной реакции свободнорадикального окисления липидов. 2,3,5-триметилгидрохинон (TMHQ) представляет собой ключевой материал для производства (dl)-токоферола и его ацетата и других производных в промышленном масштабе. Хотя описаны многочисленные способы, современное состояние уровня техники, следующее в течение прошлых 25 лет, все еще сталкивается с несколькими недостатками. В частности, производство жидких отбросов MNO2 и MNSO4 (кислая вода) увеличивает экологическую проблему. Исходя из этого, техническая проблема, формирующая основу настоящего изобретения, заключается в том, чтобы преодолеть недостатки предыдущего уровня техники и обеспечить несложный способ получения гидрохинонов, особенно такой способ, который начинается с недорогих и легко доступных материалов и достигает высоких выходов. Настоящее изобретение решает эту проблему, обеспечивая способ получения соединения гидрохинона формулы (I) в которой R2, R3, R5, R6 независимо друг от друга означает Н, А, Сус, Hal, CN, -(CYY)n-OA, -(CYY)nNYY, -O(CYY)n-OA, -O(CYY)n-NYY, -NH(CYY)n-OA или -NH(CYY)n-NYY;Y означает Н, А или Hal; А означает неразветвлнный или разветвлнный алкил, имеющий 1-10 атомов С, в котором 1-7 атомов Н могут быть замещены Hal и/или в котором одна или две соседние СН 2 группы могут быть заменены независимо друг от друга -СН=СН- и/или -CC- группой; Сус означает циклоалкил, имеющий 3-7 атомов С, в котором 1-4 атома Н могут быть заменены независимо друг от друга A, Hal и/или OY;(а) нагревания до температуры кипячения с обратным холодильником соединения фенола формулы в которой R2, R3, R5 и R6 имеют значения, как определено выше, с 0,3-0,4 экв. гексаметилентетрамина в органической кислоте, с последующим добавлением гидролитической среды и нагреванием получившейся смеси до выхода 4-гидроксибензальдегидного соединения формулы (III)(b) реакции соединения формулы (III) с окислителем в растворителе в кислых условиях до выхода соединения формулы (I). Неожиданно было продемонстрировано изобретателями, что последовательное осуществление реакции Даффа и реакции Дейкина, каждая со значительными изменениями в условиях реакции и ходе реакции, дает соответствующие гидрохиноны формулы (I) с превосходными выходами. Перед подачей этой заявки было только известно применение гексаметилентетрамина в реакции Даффа для осуществления формилирования 2,6-дизамещнных фенолов (Smith JOC 37: 3972-3973 (1972. Также было известно из ЕР 0650952 А 1, что 3,5-ди-трет-бутилсалицилальдегид может быть получен путем нагревания 2,4-дитрет-бутилфенола и 1-3 экв. гексаметилентетрамина в ледяной уксусной кислоте, добавляя 20% (об./об.) серную кислоту, и нагревания смеси снова. Способы формилирования 2,6-дизамещнных фенолов достаточно сложные, дают только средние выходы и используют дорогие, экологически-критические реактивы, такие как большие количества гексаметилентетрамина. Кроме того, окисление Дейкина 4-гидроксибензальдегидов раскрыто только при щелочных условиях, которые не подходят для синтеза гидрохинонов. Согласно предоставлению способа изобретения замещнные гидрохиноны могут быть легко получены двустадийным способом, начинающимся с формилирования замещнных фенолов до замещнного 4-гидроксибензальдегида, применяя менее чем 1 мол.экв. формильного источника углерода, предпочтительно гексаметилентетрамин, и окислением замещнного гидроксиароматического альдегида до соответствующего гидрохинона в кислых условиях, предпочтительно обеспеченных серной кислотой. Настоящее изобретение может найти применение как коммерчески привлекательный способ получения соединений формулы (I). В значении настоящего изобретения соединения формулы (I) и соединения любых других формул (II) и (III) согласно настоящему документу определены, чтобы включать их фармацевтически пригодные производные, сольваты, пролекарства, таутомеры, энантиомеры, рацематы и стереоизомеры, включая их смеси во всех соотношениях. Термин "фармацевтически пригодные производные" взят, чтобы означать, например, соли соединений согласно изобретению, а также так называемые соединения пролекарств. Термин "сольваты" соединений взят, чтобы означать продукты присоединения молекул инертных растворителей на соединения, которые сформированы вследствие их взаимной силы притяжения. Сольватами являются, например, моно- или дигидраты или алкоксиды. Термин "пролекарство" взят, чтобы означать соединения согласно изобретению, которые были изменены посредством, например, алкильной или ацильной группы, сахаров или олигопептидов и которые быстро расщепляются в организме, чтобы сформировать эффективные соединения согласно изобретению. Они также включают биоразлагаемые полимерные производные соединений согласно изобретению, как описано,например, в Int. J. Pharm. 115, 61-67 (1995). Для соединений изобретения аналогично возможно быть в форме любых желаемых пролекарств, таких как, например, сложные эфиры, карбонаты, карбаматы, мочевины, амиды или фосфаты, в случае которых фактически биологически активная форма высвобождается только через метаболизм. Любое соединение, которое может быть преобразовано in vivo, чтобы обеспечить биологически активный агент, является пролекарством в пределах объемов и идеи изобретения. В уровне техники известны различные формы пролекарств и описаны (например, Wermuth et al.,Chapter 31: 671-696, The Practice of Medicinal Chemistry, Academic Press 1996; Bundgaard, Design of Prodrugs, Elsevier 1985; Bundgaard, Chapter 5: 131-191, A Textbook of Drug Design and Development, HarwoodAcademic Publishers 1991). Упомянутые ссылки включены в данный документ посредством ссылки. Также известно, что химические вещества преобразуются в теле в метаболиты, которые могут в соответствующих случаях аналогично выявлять желаемый биологический эффект - при некоторых обстоятельствах даже в более явной форме. Любое биологически активное соединение, которое было преобразованоin vivo метаболизмом от любого из соединений изобретения является метаболитом в пределах области и идеи изобретения. Соединения изобретения могут присутствовать в форме их изомеров по двойной связи в виде чис-2 022721 тых Е- или Z-изомеров или в форме смесей этих изомеров по двойной связи. Где возможно, соединения изобретения могут быть в форме таутомеров, таких как кето-энольные таутомеры. Рассматриваются все стереоизомеры соединений изобретения, или в смеси, или в чистой, или существенно чистой форме. Соединения изобретения могут иметь асимметричные центры в любом из углеродных атомов. Следовательно, они могут существовать в форме их рацематов в форме чистых энантиомеров и/или диастереомеров или в форме смесей этих энантиомеров и/или диастереомеров. Смеси могут иметь любое желательное отношение смешения стереоизомеров. Таким образом, например, соединения изобретения, у которых есть один или больше центров хиральности и которые получают как рацематы или как диастереомерные смеси, могут фракционироваться методами, известными по сути, в их оптические чистые изомеры, то есть энантиомеры или диастереомеры. Разделение соединений изобретения может иметь место разделением на колонке на хиральные или нехиральные фазы или перекристаллизацией из необязательно оптически активного растворителя или с применением оптически активной кислоты или основания или дериватизация с оптически активным реактивом, таким как, например, оптически активный спирт, и последующее исключение радикала. Номенклатура, как применено здесь, для определения соединений, особенно соединений согласно изобретению, вообще основана на правилах IUPAC-организации для химических соединений и особенно органических соединений. Термины, обозначенные для объяснения вышеупомянутых соединений изобретения, всегда, если не обозначено иначе в описании или в формуле, имеют значения, как описано ниже. Термин "незамещнный" означает, что у соответствующего радикала, группы или фрагмента нет заместителей. Термин "замещнный" означает, что у соответствующего радикала, группы или фрагмента есть один или больше заместителей. Если у радикала есть множество заместителей, и выбор различных заместителей определен, заместители выбирают независимо друг друга и не должны быть идентичными. Даже когда у радикала есть множество отдельных определенных заместителей (например, YY), выражение такого заместителя может отличаться друг от друга (например, метил и этил). Нужно подразумевать соответственно, что многократная замена любого радикала изобретения может включать одинаковые или различные радикалы. Следовательно, если отдельные радикалы появляются неоднократно в пределах соединения, радикалы принимают обозначенные значения независимо друг от друга. Термины "алкил", "алкан" или "А" относятся к ациклическим насыщенным или ненасыщенным углеводородным радикалам, которые могут быть разветвлнными или с неразветвлнной цепью, и предпочтительно имеют 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода, то есть C1-С 10-алканилы. Примерами подходящих алкильных радикалов являются метил, этил, н-пропил, изопропил, 1,1-, 1,2- или 2,2 диметилпропил, 1-этилпропил, 1-этил-1-метилпропил, 1-этил-2-метилпропил, 1,1,2- или 1,2,2 триметилпропил, н-бутил, изобутил, втор-бутил, трет-бутил, 1-, 2- или 3-метилбутил, 1,1-, 1,2-, 1,3-, 2,2-,2,3- или 3,3-диметилбутил, 1- или 2-этилбутил, н-пентил, изопентил, неопентил, трет-пентил, 1-, 2-, 3 или -метилпентил, н-гексил, 2-гексил, изогексил, н-гептил, н-октил, н-нонил, н-децил, н-ундецил, ндодецил, н-тетрадецил, н-гексадецил, н-октадецил, н-икосанил, н-докосанил. В предпочтительном варианте осуществления изобретения "А" обозначает неразветвлнный или разветвлнный алкил, имеющий 1-10 атомов С, в котором 1-7 атомов Н могут быть замещены Hal, и/или в котором одна или две соседние СН 2 группы могут быть заменены независимо друг от друга -СН=СНи/или -СС- группой. Более предпочтительный "А" обозначает неразветвлнный или разветвлнный алкил, имеющий 1-10 атомов С, в котором 1-5 атомов Н могут быть замещены F, Cl и/или Br. Наиболее предпочтительным является C1-4-алкил. C1-4-алкильный радикал, например, представляет собой метил,этил, пропил, изопропил, бутил, изобутил, трет-бутил, втор-бутил, фторметил, дифторметил, трифторметил, пентафторэтил, 1,1,1-трифторэтил или бромметил, особенно метил, этил, пропил, изопропил, бутил или трет-бутил. Очень предпочтительным вариантом осуществления изобретения является, когда "А" обозначает метил или этил. Нужно подразумевать, что соответствующее обозначение "А" является независимым от какого-либо другого в радикалах R2, R3, R5, R6, Y и Сус. Термины "циклоалкил" или "Сус" в целях этого изобретения относятся к насыщенным и частично ненасыщенным неароматическим циклическим углеводородным группам/радикалам, имеющим 1-3 кольца, которые содержат 3-20, предпочтительно 3-12, более предпочтительно 3-9 атомов углерода. Присоединение к соединениям общей формулы (I) может происходить через любой возможный член кольца циклоалкильного радикала. Примерами подходящих циклоалкильных радикалов являются циклопропил,циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклодецил, циклогексенил, циклопентенил и циклооктадиенил. В предпочтительном варианте осуществления изобретения "Сус" обозначает циклоалкил, имеющий 3-7 атомов С, в котором 1-4 атома Н могут быть замещены независимо один от другого A, Hal и/или OY. Кроме того, определение "А" также будет включать циклоалкилы, и это должно быть применено, внося необходимые изменения, к "Сус". Термин "алкилокси" или "алкокси" в целях этого изобретения относится к алкильному радикалу,соответствующему вышеупомянутому определению, который присоединен к атому кислорода. Присоединение к соединениям общей формулы (I) состоит через атом кислорода. Примерами являются метокси,этокси и н-пропилокси, пропокси, изопропокси и бутокси. Предпочтительным является "С 1-С 4-3 022721 алкилокси", имеющий указанное число атомов углерода. Термин "галоген", "атом галогена", "галогеновый заместитель" или "Hal" в целях этого изобретения относится к одному или, где необходимо, множеству атомов фтора (F, фтор), брома (Br, бром), хлора (Cl,хлор) или йода (I, йод). Обозначения "дигалоген", "тригалоген" и "пергалоген" относятся, соответственно, к двум, трем и четырем заместителям, где каждый заместитель может быть независимо выбран из группы, включающей фтор, хлор, бром и йод. "Галоген" предпочтительно означает атом фтора, хлора или брома. Фтор и хлор являются более предпочтительными, когда галогены замещены алкильной (галоалкильной) или алкоксигруппой (например, CF3 и CF3O). Термин "гидроксил" означает -ОН группу. Индекс n предпочтительно равен 0, 1 или 2. Хотя родовая формула (I) может даже быть определена настолько же широко, как в ЕР 0599148 В 1 в одном варианте осуществления, здесь другой вариант осуществления изобретения включает способ получения соединений формулы (I), в которойR2, R3, R5, R6 независимо друг от друга означают Н, A, Hal или ОА; А означает неразветвлнный или разветвлнный алкил, имеющий 1-10 атомов С, в котором 1-5 атомов Н могут быть замещены Hal; иHal означает F, Cl или Br. В предпочтительном варианте осуществления изобретения получают соединения формулы (I), в которойR6 означает Н; А означает неразветвлнный или разветвлнный алкил, имеющий 1-4 атомов С; иHal означает F, Cl или Br. Должно быть понято, что соединения формулы (I) предпочтительно являются ди- или тризамещнными в этом документе ниже. В этом аспекте радикалы R3 и R5 могут быть либо одинаковыми, либо разными, но они предпочтительно являются одинаковыми. В более предпочтительном варианте осуществления изобретения получают соединения формулыHal означает F, Cl или Br. В наиболее предпочтительном варианте осуществления изобретения получают соединения формулы (I), в которойR3, R5 означают метил или этил;R6 означает Н. В очень предпочтительном варианте осуществления изобретения получают 2,3,5-триметилбензол 1,4-диол и/или 2,6-диэтилбензол-1,4-диол. Само собой разумеется, что 2,3,5-триметилбензол-1,4-диол соответствует 2,3,5-триметилгидрохинону (TMHQ). Располагающим стартовым материалом для производства TMHQ согласно изобретению является 2,3,6-триметилфенол, который легко доступен коммерческим путем. Промежуточное соединение 2,3,5-триметил-4-гидроксибензальдегид не было получено ранее до раскрытия этого изобретения. Его синтезируют на стадии (а) способа изобретения и характеризуют ниже. Даже при том, что стартовые материалы не являются коммерчески доступными, они могут быть произведены методами, известными по сути, как описано в литературе (например, в стандартных работах, таких как Houben-Weyl, Methoden der organischen Chemie [Methods of Organic Chemistry], GeorgThieme-Verlag, Stuttgart), то есть при условиях реакций, которые известны и подходят упомянутым реакциям. При желании стартовые материалы могут также быть образованы in-situ, оставляя их в невыделенном состоянии в сырой смеси реакции, но немедленно преобразовывая их далее в соединение согласно изобретению. С другой стороны, возможно выполнение постадийной реакции. Настоящее изобретение раскрыто, чтобы приспособить условия реакции Даффа и реакции Дейкина,чтобы получить желаемые высокие выходы продукта гидрохинона. В реакции на стадии (а), гексаметилентетрамин применяют в количестве 0,3-0,4 мол.экв. гексаметилентетрамина без какой-либо существенной потери выхода продукта. Должно быть понято, что "эквивалент" обозначает молярное соотношение гексаметилентетрамина к соединению формулы (II). Указанная реакция с соединением фенола формулы (II) осуществляется в органической кислоте, такой как карбоновая кислота или сульфоновая кислота; пригодность которой для формилирования может быть вычислена специалистом в данной области обычным способом. Несколько общих примеров включают молочную кислоту, уксусную кислоту, муравьиную кислоту, лимонную кислоту, щавелевую кислоту и мочевую кислоту. В предпочтительном аспекте изобретения стадия (а) способа осуществляется в уксусной кислоте. Более удобно применять ледяную уксусную кислоту, особенно в диапазоне 98-100%(об./об.), чтобы максимизировать выход наряду с минимальным формированием примеси. Применение восстановленной уксусной кислоты дает тот же самый выход. Смесь соединения фенола формулы (II) и гексаметилентетрамина в органической кислоте нагревают на протяжении интервала времени 1-5 ч, предпочтительно 2-3,5 ч. Применение повышенной температуры желательно, чтобы получить гомогенные условия реакции, а потому температура кипячения с обратным холодильником, особенно при атмосферном давлении, наиболее удобна и эффективна и поэтому предпочтительна в диапазоне 90-135 С, очень предпочтительно в диапазоне 110-130 С. За периодом нагревания на стадии (а) следует быстрое охлаждение гидролитической средой в количестве, примерно равном объему органической кислоты, который применяется (применяемая органическая кислота, например уксусная кислота). Гидролитическая среда предпочтительно представляет собой воду или водный раствор кислоты, более предпочтительно серной кислоты, наиболее предпочтительно 10-20% (об./об.) серной кислоты. Должно быть понято, что специалист в данной области знает несколько других типов водных неорганических кислот, которые подходят в пределах настоящего изобретения,такие как HCl, Н 2 СО 3, Н 3 РО 4 и т.п. Водную смесь далее нагревают в течение короткого периода времени,в частности от 0,25 до 1 ч. Нагревание осуществляется при температурных условиях, как дано выше, но очень предпочтительно в диапазоне 100-120 С. Реакцию стадии (а) останавливают охлаждением и концентрируют сырое промежуточное соединение формулы (III), таким способом, как фильтрация, промывают и сушат до выходов по меньшей мере 60%, предпочтительно по меньшей мере 70%, более предпочтительно по меньшей мере 80%, наиболее предпочтительно по меньшей мере 90%. Альтернативно, реакционный раствор может быть сконцентрирован под вакуумом, чтобы регенерировать органическую кислоту, или охлажден и экстрагирован неполярным растворителем. Предпочтительными растворителями для экстракции являются углеводородные растворители, такие как гексан, гептан, толуол и ксилол, или простые эфиры, такие как диэтиловый эфир и трет-бутилметиловый эфир. Перед стадией (b) соединение формулы (III) может быть очищено, например, путем пропускания раствора в неполярном растворителе, таком как гексан или толуол, через слой силикагеля, и растворитель отслаивают из фильтрата, чтобы обеспечить твердое вещество. Альтернативно, органическая фаза может быть отделена от водной фазы, и органическую фазу затем перекристаллизовывают в спиртовом растворителе, особенно метаноле. Предпочтительная последовательная переработка осуществляется при помощи неорганического основания, более предпочтительно гидроксида натрия, наиболее предпочтительно 0,1-10% гидроксида натрия в молярном соотношении соединения формулы (III) к гидроксиду натрия от 0,2 до 5. Очень предпочтительно применяется 1-2% гидроксида натрия в эквимолярном соотношении соединения формулы (III) к гидроксиду натрия, чтобы увеличить чистоту, в то время как выходы в основном держатся в пределах предпочтительных диапазонов, как дано выше. В окислении Дейкина стадии (b) соединение формулы (III) реагирует с окислителем, таким как нитрат, хлорит, хлорат, перхлорат и другие аналогичные галогеновые соединения аммоний церия(IV), соединения шестивалентного хрома (например, хромовая и двухромовая кислоты и триоксид хрома, хлорхромат пиридиния), хромат/дихроматные соединения, гипохлорит и другие гипогалитсоединения, йод и другие галогены, азотная кислота, оксид азота, осмиевая четырехокись, озон, соли перманганата, пероксидные соединения, надсерная кислота, нитрат калия или сульфоксиды. Предпочтительным окислителем изобретения является пероксид водорода, более предпочтительно 20-50% пероксид водорода, наиболее предпочтительно 30% пероксид водорода, очень предпочтительно приблизительно 1,2 мол.экв. 30% пероксида водорода. Реакция в основном проводится в инертном растворителе. Подходящими инертными растворителями являются, например, углеводороды, такие как гексан, петролейный эфир, бензол, толуол или ксилол; хлорированные углеводороды, такие как трихлорэтилен, 1,2-дихлорэтан, четыреххлористый углерод,хлороформ или дихлорметан; спирты, такие как метанол, этанол, изопропанол, н-пропанол, н-бутанол или трет-бутанол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран(THF) или диоксан; гликолевые простые эфиры, такие как монометиловый или моноэтиловый эфир этиленгликоля, диметиловый эфир этиленгликоля (диглим); кетоны, такие как ацетон или бутанон; амиды,такие как ацетамид, диметилацетамид или диметилформамид (DMF); нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид (DMSO); дисульфид углерода; карбоновые кислоты, такие как муравьиная кислота или уксусная кислота; нитросоединения, такие как нитрометан или нитробензол; сложные эфиры, такие как этилацетат, или смеси указанных растворителей. Предпочтение отдают воде или органическим растворителям. Более предпочтительно применять алкан или спирт, более предпочтительно метанол, наиболее предпочтительно 98-100% чистый метанол. Кроме этого, реакция по существу проводится в кислых условиях. Это относится к органическим и неорганическим кислотам, уже упомянутым в ходе предыдущего описания как подходящие примеры. На стадии (b) кислотные условия предпочтительно обеспечены серной кислотой. Количество применяемой серной кислоты более предпочтительно составляет около 0,1-0,3 мол.экв., но лучшие результаты получены при применении 0,2 мол.экв. серной кислоты. Другими словами, более предпочтительно применять серную кислоту в молярном соотношении к соединению формулы (III) от 0,1 до 0,3, наиболее предпочтительно между 0,1 и 0,3, очень предпочтительно серная кислота в молярном соотношении к соединению формулы (III) 0,2. Чистота серной кислоты должна составлять особенно по меньшей мере 98%. Должно быть понято, что пороговые значения не покрываются, определяя диапазон "между". Реакция Дейкина предпочтительно осуществляется при температуре между 15 и 90 С. Температурный диапазон между 15 и 50 С еще более желателен, чтобы получить гомогенные условия реакции. Температура может измениться во время окисления, которое выполняется на протяжении интервала времени 0,5-2 ч. После завершения окисления Дейкина избыток пероксида водорода и образованный хинон путем переокисления гидрохинона могут быть удалены. Для возвращающего удаления предпочтителен восстанавливающий агент. Общепринятыми восстанавливающими агентами являются ион двухвалентного железа, алюмогидрид лития, водород в момент выделения, амальгама натрия, борогидрит натрия, сульфитные соединения, гидразин, цинк-ртутная амальгама, диизобутилалюминий гидрит, щавелевая кислота или муравьиная кислота. Более предпочтительно применять дитионат натрия, наиболее предпочтительно 10-20% раствор дитионата натрия в воде. Избыточный пероксид водорода может также быть удален возвращающим образом путем добавления NaHSO3, такого как 10% водный раствор NaHSO3 в особенности. Возвращающее воздействие при комнатной температуре проходит от 0,5 до 1 ч. Далее растворитель дистиллирован под вакуумом. Восстановление метанола до 60-75% является предпочтительным, чтобы улучшить выход. Дистилляцию растворителя останавливают охлаждением и гидрохинон формулы (I) концентрируют, например, фильтрацией, промывают и сушат до выходов по меньшей мере 60%, предпочтительно по меньшей мере 70%, более предпочтительно по меньшей мере 80%, очень предпочтительно по меньшей мере 85%. В частности, получена высокая чистота более, чем 97% для конечного продукта гидрохинона. Необязательно представлена соль соединений согласно формул (I)-(III), предпочтительно формулы(I). Соединения согласно изобретению могут быть применены в их заключительной несолевой форме. С другой стороны, настоящее изобретение также охватывает применение этих соединений в форме их фармацевтически приемлемых солей, которые могут быть получены из различных органических и неорганических кислот и оснований процедурами, известными в уровне техники. Фармацевтически приемлемые солевые формы соединений согласно изобретению по большей части получены обычными методами. Относительно вышеизложенного можно заметить, что выражения "фармацевтически приемлемая соль" и "физиологически приемлемая соль", которые здесь используются взаимозаменяемыми, в существующей связи взяты, чтобы означать активный ингредиент, который включает соединение согласно изобретению в форме одной из его солей, в особенности если эта солевая форма передает улучшенные фармакокинетические свойства на активном ингредиенте по сравнению со свободной формой активного ингредиента или какой-либо другой солевой формой активного ингредиента, используемой ранее. Фармацевтически приемлемая солевая форма активного ингредиента может также предоставить этому активному ингредиенту впервые желаемую фармакокинетическую особенность, которую он не имел ранее и может даже иметь положительное влияние на фармакодинамику этого активного ингредиента относительно его терапевтической эффективности в теле. В объеме настоящего изобретения был обеспечен многообещающий новый подход для производства гидрохинонов формулы (I). Синтез гидрохинона включает вставку и удаление одного атома углерода,который родственный с природой и экологически неопасный. Способ имеет две стадии, начиная от замещнных фенолов формулы (II). Применение дешевого сырья имеет выгоду для экономичного поведения способа. Ключевые промежуточные соединения замещнных гидроксиароматических альдегидов формулы (III) выгодно выделены в чистом состоянии и охарактеризованы спектральными данными. Кроме того, примеси, образованные в процессе, полностью характеризуются спектроскопическими методами, и их устранение в промежуточном и конечном продукте хорошо изучено. Соответственно, этот способ обладает общей применимостью для преобразования различных замещнных фенолов формулы(II) до замещнных гидрохинонов формулы (I) через соответствующие замещнные альдегиды формулы(III). На практике изобретения новый способ делает возможным получение TMHQ, начиная от 2,3,6 триметилфенола. Конечный продукт получают с высокими выходами и высокой чистотой (97%). Все ссылки, процитированные здесь, таким образом, включены посредством ссылок в раскрытии изобретения. Нужно подразумевать, что это изобретение не ограничено отдельными соединениями, применениями и способами, описанными здесь, объект как таковой может, конечно, изменяться. Нужно также подразумевать, что терминология, используемая здесь, приведена только с целью описания отдельных вариантов осуществления и не предназначена, чтобы ограничить область данного изобретения, которая определена только приложенной формулой изобретения. Как применено здесь, включая приложенную формулу изобретения, формы слов в единственном числе включают их соответствующие множественные референты, если контекст ясно не указывает иначе. Таким образом, например, ссылка на "соединение" включает одно или несколько различных соединений, и ссылка на "способ" включает ссылку на эквивалентные стадии и методы, известные специалисту в данной области техники и т.д. Если иначе не определено, все технические и научные термины, использованные здесь, имеют те же значения, которые обычно понимаются специалистом в данной области техники, которой принадлежит это изобретение. Методы, которые существенны согласно изобретению, изложены подробно в описании. Другие методы, которые не описаны подробно, соответствуют известным стандартным методам, которые известны специалисту в данной области техники, или методы описаны более подробно в процитированных ссылках, заявках на патент или стандартной литературе. Хотя в практике или тестировании настоящего изобретения могут применяться методы и материалы, подобные или эквивалентные описанным здесь, подходящие примеры описаны ниже. Следующие примеры обеспечены посредством иллюстрации, а не посредством ограничения. В пределах примеров применяются стандартные реактивы и буфера, которые не имеют загрязняющих действий (всякий раз на практике). Пример особенно должен быть понят таким образом, что он не ограничивается явно продемонстрированными комбинациями признаков, но иллюстрируемые признаки могут неограниченно сочетаться снова, если решается техническая проблема изобретения. Пример 1. Синтез 2,6-дихлорбензол-1,4-диола. В 250 мл трехгорлую круглодонную колбу, оснащенную обратным холодильником, термометром для колб и механической мешалкой, загружали 10 г (0,061 моль) 2,6-дихлорфенола, 40 мл ледяной уксусной кислоты и 3,44 г (0,024 моль) гексаметилентетрамина. Реакционную массу нагревали при кипячении с обратным холодильником на протяжении 3 ч (прозрачный раствор был получен при 70 С). Ход реакции проверяли ТСХ. Смеси позволяли остыть до 100 С; медленно добавляли 30 мл 10% серной кислоты и перемешивали при 100 С на протяжении 20-30 мин. Реакционную массу выливали в 200 г измельченного льда. Твердое вещество отфильтровывали и хорошо промывали 100 мл воды и сушили при 70 С, при этом получали 9,2 г (84%) 3,5-дихлор-4-гидроксибензальдегида. Температура плавления: 153155 С. 1 Н ЯМР (400 МГц, CD3OD)9.72 (s, 1H),7.80 (s, 1H). В 250 мл круглодонной колбе готовили раствор 0,69 г (0,007 моль) серной кислоты и 40 мл метанола. Добавляли 4,5 г (0,023 моль) 3,5-дихлор-4-гидроксилбензальдегида и перемешивали до получения прозрачного раствора. Реакционную массу охлаждали до 17-20 С и при перемешивании медленно добавляли 5,34 г (0,047 моль) 30% раствора пероксида водорода (внимание: экзотермическая реакция) на протяжении периода 5-10 мин при поддержании температуры 20-35 С. Смесь перемешивали на протяжении 30 мин при комнатной температуре и при 80 С на протяжении 60 мин. Завершение реакции подтверждали ТСХ. При перемешивании медленно добавляли 15 мл 20% раствора дитионита натрия (5-10 мин) при 20-25 С. Смесь перемешивали под атмосферой азота при 20-25 С на протяжении 30 мин. 20 мл водного раствора метанола дистиллировали при 40-50 С. Реакционную массу охлаждали до 20-25 С и на протяжении периода 5 мин добавляли 45 мл технической воды. Смесь перемешивали 10-15 мин под атмосферой азота. Продукт отфильтровывали и промывали 25 мл технической воды, хорошо отжимали и затем сушили под вакуумом при 60 С на протяжении 2 ч, чтобы получить 3,2 г (76%) 2,6-дихлоргидрохинона(2,6-дихлорбензол-1,4-диола). Температура плавления: 110-112 С. Пример 2 А. Синтез 2,3,5-триметилбензол-1,4-диола. Смесь 2,3,6-триметилфенола (5 г, 0,036 моль) и гексаметилентетрамина (2,06 г, 0,0147 моль) в уксусной кислоте (20 мл) перемешивали при 115 С на протяжении 2 ч. Растворитель (10 мл) дистиллировали и добавляли 20% (об./об.) H2SO4 (20 мл) и затем смесь перемешивали при 115 С на протяжении 20 мин. Смесь затем выливали в ледяную холодную воду (50 мл) и отделившееся твердое вещество отфильтровывали, хорошо промывали холодной водой (25 мл) и отжимали под вакуумом. Влажный осадок сушили при 90 С на протяжении 2 ч под вакуумом до выхода 2,3,5-триметил-4-гидроксибензальдегида(4,5 г, 75%; табл. 1,12). Готовили раствор метанола (40 мл) и 0,267 г (0,0027 моль) серной кислоты и перемешивали. В этот раствор добавляли 4-гидрокси-2,3,5-триметилбензальдегида (4,5 г, 0,027 моль) и перемешивали до растворения. 30% пероксид водорода (3,46 г, 0,029 моль) медленно добавляли к этому прозрачному раствору на протяжении 5 мин. После завершения добавления реакционную смесь перемешивали при комнатной температуре на протяжении 30 мин. Растворитель (20 мл) выпаривали под вакуумом и под атмосферой азота добавляли 50 мл 20% раствора дитионита натрия. Реакционную массу перемешивали на протяжении 30 мин при 20-25 С, фильтровали, промывали холодной водой, хорошо отжимали и, следовательно, хорошо сушили при 80 С под вакуумом на протяжении 2 ч до выхода 2,3,5-триметилгидрохинона (2,3,5-триметилбензол-1,4-диол) в виде тускло-жлтого твердого вещества (3,33 г, 80%; табл. 2, 13). Пример 2 В. Синтез 2,3,5-триметилбензол-1,4-диола. В 500 мл трехгорлую круглодонную колбу, оборудованную механической мешалкой и термометром для колб, загружали 50 г (0,36 моль) 2,3,6-триметилфенола, 200 мл ледяной уксусной кислоты и 20,61 г(0,14 моль) гексаметилентетрамина. Реакционную массу кипятили с обратным холодильником на протяжении 2 ч при 120-130 С. Завершение реакции подтверждали ТСХ. 100 мл уксусной кислоты дистиллировали при температуре кипячения с обратным холодильником. Реакционной массе позволяли остыть при 100 С и на протяжении периода 5-10 мин медленно добавляли 50 мл 20% серной кислоты. Смесь перемешивали при 100 С 15-30 мин. Нагревание останавливали и реакционную массу охлаждали до 20 С в ледяной бане. Добавляли 500 мл технической воды и перемешивали 10 мин при 20-25 С. Реакционную массу отфильтровывали и промывали 300 мл технической воды. Сырой продукт сушили при 60-80 С под вакуумом, чтобы получить 54 г (90%) сырого 2,3,5-триметил-4-гидроксибензальдегида. Далее готовили 1% один эквивалентный раствор гидроксида натрия в технической воде. К нему добавляли 54 г сырого 2,3,5-триметил-4-гидроксибензальдегида при перемешивании при комнатной температуре. Смесь перемешивали 15-20 мин и фильтровали. Остаток должен был быть соскоблен как примесь. Фильтрат собирали, подкисляли 19,75 г ледяной уксусной кислоты и фильтровали. Продукт отжимали под вакуумом,сушили при 60-80 С на протяжении 3-4 ч до получения 48 г (80%) чистого 2,3,5-триметил-4-гидроксибензальдегида в виде грязно-белого аморфного вещества (табл. 1,13). Температура плавления: 135137 С. 1 Н ЯМР (400 МГц, CDCl3)10.15 (s, 1H),7.49 (s, 1H),5.32 (bs, 1H),2.58 (s, 3H),2.28 (s,3 Н),2.22 (s, 3 Н). В 500 мл круглодонной колбе готовили раствор 5,73 г (0,058 моль) серной кислоты и 432 мл метанола. 48 г (0,29 моль) очищенного 2,3,5-триметил-4-гидроксибензальдегида добавляли при перемешивании при комнатной температуре. Смесь дополнительно перемешивали 10-15 мин до получения прозрачного раствора. Реакционную массу охлаждали до 17-20 С и 38,09 г (0,33 моль) 30% раствора пероксида водорода медленно добавляли при перемешивании (внимание: экзотермическая реакция) на протяжении периода 20-30 мин, поддерживая температуру 20-35 С. Смесь перемешивали на протяжении 30 мин при комнатной температуре и завершение реакции было подтверждено ТСХ. 58 мл 20% раствора дитионита натрия медленно добавляли (15-20 мин) при перемешивании при 20-25 С. Смесь перемешивали под атмосферой азота при 20-25 С на протяжении 30 мин. Из реакционной массы при 40-50 С дистиллировали 220 мл водного раствора метанола. Реакционную массу охлаждали до 20-25 С и добавляли 480 мл технической воды на протяжении периода 15-20 мин. Смесь перемешивали 10-15 мин под атмосферой азота. Продукт отфильтровывали и промывали 250 мл технической воды, хорошо отжимали и сушили под вакуумом при 60 С на протяжении 2 ч до получения 37,8 г (85%) 2,3,6-триметилгидрохинона (2,3,5 триметилбензол-1,4-диол) в виде тускло-жлтого твердого вещества (табл. 2,14). Температура плавления: 172-174 С. Пример 2 С. Переменные условия реакции формилирования в примере 2A и 2 В. Формилирование 2,3,6-триметилфенола первоначально осуществляли, изменяя условия реакции. Детали, перечисленные в табл. 1,1, 2 и 11, рассматривают как сравнительные примеры и не рассматривают под объемом настоящего изобретения. Таблица 1 Условия реакции формилирования Пример 2D. Формилирование 2,3,6-триметилфенола формальдегидом и ацетатом аммония. Вместо гексаметилентетрамина, как применялось в примере 2 А и 2 В, для формилирования были применены формальдегид и ацетат аммония в уксусной кислоте, как проиллюстрировано ниже. В 250 мл трехгорлую круглодонную колбу, оборудованную обратным холодильником, загружали 20,4 г (0,15 моль) 2,3,6-триметилфенола. Туда добавляли 100 мл уксусной кислоты, 3,09 г (0,04 моль) ацетата аммония и 3,243 г (0,04 моль) раствора формалина. Реакционную массу нагревали при 120 С на протяжении периода 2,5 ч. Завершение реакции подтверждали ТСХ. Реакционную массу выливали в 400 мл ледяной холодной воды при перемешивании. Твердое вещество отфильтровывали и промывали 250 мл технической воды. Продукт сушили под вакуумом до получения 17 г (75%) сырого 2,3,5-триметил-4-гидроксибензальдегида. Очищали сырой продукт 1% раствором гидроксида натрия при этом получали 13 г (53%) чистого 2,3,5-триметил-4-гидроксилбензальдегида. Пример 2 Е. Переменные условия окисления Дейкина в примере 2 А и 2 В. Реакцию Дейкина 2,3,5-триметил-4-гидроксибензальдегида первоначально осуществляли, изменяя условия реакции. Детали перечислены в табл. 2. Номера 1-12 рассматривают как сравнительные примеры и не рассматривают под объемом настоящего изобретения. Таблица 2 Условия окисления ДейкинаDCM - дихлорметан Пример 3. Синтез далее замещнных гидрохинонов формулы (I). Методология этого изобретения была расширена на производство других ингибиторов окисления,применяемых теперь. В соответствии с примером 2 В соединения табл. 3,3 и 4 были приготовлены,используя молярное соотношение замещнного фенола к гексаметилентетрамину 1:0,4 и ледяную уксусную кислоту (550 мл органического основания на 1 моль замещнного фенола). Время кипячения с обратным холодильником было 2-3 ч. Таблица 3 Краткие характеристики соединений примеров 1-4 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения гидрохинона формулы (I) в которой R2, R3, R5, R6 независимо друг от друга означают Н, А, Сус, Hal, CN, -(CYY)n-OA, -(CYY)nNYY, -O(CYY)n-OA, -O(CYY)n-NYY, -NH(CYY)n-OA или -NH(CYY)n-NYY;Y означает Н, А или Hal; А означает неразветвлнный или разветвлнный алкил, имеющий 1-10 атомов С, в котором 1-7 атомов Н могут быть замещены Hal и/или в котором одна или две соседние СН 2 группы могут быть заменены независимо друг от друга -СН=СН- и/или -СС- группой; Сус означает циклоалкил, имеющий 3-7 атомов С, в котором 1-4 атома Н могут быть заменены независимо друг от друга A, Hal и/или OY;(а) нагревания до температуры кипячения с обратным холодильником соединения фенола формулы в которой R2, R3, R5 и R6 имеют значения, как определено выше, с 0,3-0,4 экв. гексаметилентетрамина в органической кислоте, с последующим добавлением гидролитической среды и нагреванием получившейся смеси до получения 4-гидроксибензальдегидного соединения формулы (III)(b) реакции соединения формулы (III) с окислителем в растворителе в кислых условиях до получения соединения формулы (I). 2. Способ по п.1 получения соединения формулы (I), в которойHal означает F, Cl или Br. 3. Способ по п.2 для получения 2,3,5-триметилбензол-1,4-диола и/или 2,6-диэтилбензол-1,4-диола. 4. Способ по любому из пп.1-3, в котором на стадии (а) органической кислотой является ледяная уксусная кислота и/или гидролитическая среда представляет собой воду или серную кислоту. 5. Способ по любому из пп.1-4, в котором на стадии (а) нагревание осуществляют при температуре кипячения с обратным холодильником 110-130 С и/или на стадии (b) реакцию проводят при температуре между 15 и 50 С. 6. Способ по любому из пп.1-5, в котором на стадии (b) окислитель представляет собой пероксид водорода и/или растворителем является спирт. 7. Способ по любому из пп.1-6, в котором на стадии (b) кислотные условия обеспечивают серной кислотой в молярном соотношении к соединению формулы (III), составляющем 0,2. 8. Способ по любому из пп.1-7, в котором перед стадией (b) соединение формулы (III) очищают 0,110% гидроксидом натрия в молярном соотношении соединения формулы (III) к гидроксиду натрия от 0,2 до 5. 9. Способ по п.8, в котором перед стадией (b) соединение формулы (III) очищают 1-2% гидроксидом натрия в эквимолярном соотношении соединения формулы (III) к гидроксиду натрия. 10. Способ по любому из пп.1-9, в котором после стадии (b) соединение формулы (I) очищают 1020% раствором дитионата натрия.
МПК / Метки
МПК: C07C 45/36, C07C 37/56, C07C 39/30, C07C 47/542, C07C 39/08
Метки: получения, способ, гидрохинонов
Код ссылки
<a href="https://eas.patents.su/11-22721-sposob-polucheniya-gidrohinonov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения гидрохинонов</a>
Способ получения дихлорпропанола, способ получения эпихлоргидрина, способ получения эпоксидных смол и применение оборудования, обладающего коррозионной стойкостью, в способе получения дихлорпропанола

Номер патента: 14241
Опубликовано: 29.10.2010
Авторы: Франк Кристиан, Краффт Филипп, Жильбо Патрик, Сметс Валентин, Бальтазар Доминик
МПК: C07C 29/62, C07C 31/36, B01J 19/02...
Метки: оборудования, применение, способ, стойкостью, способе, смол, эпихлоргидрина, обладающего, эпоксидных, получения, дихлорпропанола, коррозионной
Формула / Реферат:
1.Способ получения дихлорпропанола, содержащий:(a) стадию, на которой глицерин или сложный эфир глицерина или их смесь вводят во взаимодействие с агентом хлорирования, содержащим хлороводород,(b) по меньшей мере одну другую стадию, осуществляемую на оборудовании, выполненном или имеющем покрытие из материалов, обладающих стойкостью по отношению к агенту хлорирования, в условиях осуществления этой стадии,причем другая стадия является стадией...
Цеолитный катализатор l-типа, способ его получения, способ получения ароматических углеводородов, способ получения бензина

Номер патента: 3559
Опубликовано: 26.06.2003
Авторы: Фукунага Тецуя, Иннес Роберт А., Сугимото Митио
МПК: C10G 35/095, B01J 29/61, C07C 5/41...
Метки: l-типа, ароматических, получения, цеолитный, катализатор, углеводородов, способ, бензина
Формула / Реферат:
1. Цеолитный катализатор L-типа, который получают при нанесении на цеолит L-типа платинового компонента, одного или более галогеновых компонентов и одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, при этом наносимое количество одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, находится в интервале от 0,001 до 3 мас.% из расчета на общую массу катализатора, молярное отношение...
Способ получения полиолефина, применение смеси по меньшей мере двух агентов переноса цепи для получения полиолефина, полиэтилен низкой плотности, способ получения кабеля и кабель

Номер патента: 19226
Опубликовано: 28.02.2014
Авторы: Воигт Бьерн, Смедберг Анника, Хубер Маркус, Нильссон Ульф, Кампус Альфред, Шильд Герман
МПК: C08F 10/02, C08F 2/38, C08L 23/06...
Метки: полиэтилен, мере, двух, применение, получения, кабель, цепи, меньшей, полиолефина, переноса, агентов, смеси, кабеля, способ, низкой, плотности
Формула / Реферат:
1. Способ получения полиолефина, включающий полимеризацию по меньшей мере одного олефина в присутствии смеси по меньшей мере двух агентов переноса цепи, при этом смесь включаетполярный агент переноса цепи (полярный АПЦ) инеполярный агент переноса цепи (неполярный АПЦ), который представляет собой неароматический линейный, разветвленный или циклический углеводород, возможно содержащий гетероатом, такой как О, N, S, Si или Р, и не содержит полярной...
Катализатор для получения сложных эфиров,способ получения сложного эфира и способ получения сложного полиэфира с участием такого катализатора

Номер патента: 11171
Опубликовано: 27.02.2009
Авторы: Макинтош Кэлам Гарри, Партридж Мартин Грэхэм, Хэнратти Алан Джозеф
МПК: C08G 63/85, B01J 31/04
Метки: участием, сложных, катализатора, сложного, эфиров,способ, катализатор, способ, эфира, такого, получения, полиэфира
Формула / Реферат:
1. Катализатор для получения сложного эфира в реакции этерификации, состоящий из продукта взаимодействия: a) соединения титана, циркония или гафния; b) 2-оксикарбоновой кислоты; c) четвертичного аммониевого соединения, выбранного из группы, состоящей из гидроксида тетраэтиламмония и гидроксида тетраметиламмония, и d) соединения цинка. 2. Катализатор по п. 1, в котором соединением титана, циркония или гафния является алкоголят, имеющий формулу...
Способ получения дихлорпропанола, способ получения эпихлоргидрина и способ получения эпоксидных смол

Номер патента: 13681
Опубликовано: 30.06.2010
Авторы: Жильбо Патрик, Краффт Филипп
МПК: C07C 31/36, C07C 31/42, C07C 29/62...
Метки: дихлорпропанола, способ, получения, смол, эпихлоргидрина, эпоксидных
Формула / Реферат:
1. Способ получения дихлорпропанола, в котором вводят во взаимодействие глицерин, или сложный эфир глицерина, или их смесь, общее содержание металлов в которых, выраженное в расчете на элементы, выше или равно 0,1 мкг/кг и ниже или равно 1000 мг/кг, и агент хлорирования.2. Способ по п.1, в котором общее содержание металлов ниже или равно 500 мг/кг и который характеризуется по меньшей мере одним из следующих признаков:содержание железа в...
Предыдущий патент: Огнеупорный продукт с высоким содержанием диоксида циркония
Следующий патент: Подложка, снабженная набором с термическими свойствами, в частности, для реализации обогреваемого стекла
Случайный патент: Фанера и способ ее изготовления