Насыпной катализатор
Номер патента: 12365
Опубликовано: 30.10.2009
Авторы: Сабато Мисео, Леливелд Робертус Герардис, Плантенга Франс Л., Солед Стюарт Леон, Эйсбаутс-Шпичкова Сонья, Лаувен Якобус Николас













Формула / Реферат
1. Насыпной катализатор, включающий по меньшей мере 60 мас.% частиц оксидов металлов, содержащих один или более металлов группы VIII и молибден, включающий менее чем 10 мол.% любого другого металла группы VIB относительно суммарного количества металлов группы VIB и металл группы V в количестве менее чем 10 мол.% относительно суммы металлов группы VIB, где насыпной катализатор прокален при температуре ниже температуры, при которой гексагональная метастабильная кристаллическая структура превращается в неактивную кристаллическую структуру, и где насыпной катализатор имеет метастабильную гексагональную фазу, характеризуемую рентгенограммой, имеющей отражения при 33-35 и 58-61ш2q.
2. Насыпной катализатор по п.1, в котором молярное отношение металла группы VIII к металлу группы VIB составляет более 1,5.
3. Насыпной катализатор по п.1, в котором молярное отношение металла группы VIII к металлу группы VIB составляет от 2,5 до 5.
4. Насыпной катализатор по п.1, у которого главные отражения имеют полную ширину на половине максимума менее чем 2,5.
5. Насыпной катализатор по п.1, в котором металлом группы V является ниобий.
6. Насыпной катализатор по п.1, в котором частицы оксидов металлов практически включают только один металл группы VIII.
7. Способ получения насыпного катализатора по п.1, включающий:
(i) приготовление реакционной смеси, состоящей из протонной жидкости, одного или более соединений первого металла, включающего один или более металл группы VIII, и соединения, включающего молибден, и меньше чем 10 мол.% любого другого металла группы VIB относительно суммарного количества металлов группы VIB и/или металла группы V в количестве меньше чем 10 мол.% относительно суммы металлов группы VIB,
(ii) осуществление взаимодействия соединений первого и второго металлов при повышенной температуре, при котором твердые соединения первого и/или второго металла остаются, по меньшей мере, частично в твердом состоянии в процессе всей реакции с образованием частиц оксидов металлов,
где насыпной катализатор на всех стадиях в ходе его приготовления остается при температуре ниже температуры, при которой гексагональная метастабильная кристаллическая структура превращается в неактивную кристаллическую структуру.
8. Способ по п.7, в котором соединение первого металла и соединение второго металла остается, по меньшей мере, частично в твердом состоянии в процессе всей реакции.
9. Способ по п.8, в котором соединением первого металла является гидроксикарбонатом металла и соединением второго металла является оксид металла или кислота.
10. Способ по п.9, в котором соединением первого металла является карбонат или гидроксикарбонат никеля, имеющий площадь поверхности по меньшей мере 150 м2/г.
11. Способ получения насыпного катализатора по п.1, включающий:
(i) приготовление раствора одного или более растворимых соединений первого металла, включающих один или более металлов группы VIII, и растворимого соединения второго металла, включающего молибден, и менее чем 10 мол.% относительно суммарного количества металлов группы VIB и/или металл группы V в количестве менее чем 10 мол.% относительно суммы металлов группы VIB,
(ii) осуществление взаимодействия и/или осаждение соединений первого и второго металлов с образованием частиц оксидов,
где насыпной катализатор на всех стадиях в процессе его приготовления остается при температуре ниже температуры, при которой гексагональная метастабильная кристаллическая структура превращается в неактивную кристаллическую структуру.
12. Способ по п.7, в котором протонной жидкостью является вода, и реакцию проводят при гидротермальных условиях при температуре реакции выше температуры кипения протонной жидкости.
13. Способ по п.7, в котором твердые соединения первого и второго металлов не содержат атомов азота и в котором протонную жидкость, отделенную от прореагировавших частиц оксидов металлов на стадии iii, повторно используют, по меньшей мере, частично для получения суспензии на стадии i.
14. Способ по п.7, дополнительно включающий одну или более из следующих стадий способа:
(iii) отделения частиц оксидов металлов от реакционной смеси,
(iv) необязательное смешение частиц оксидов металлов с от 0 до 40 мас.% одного или более материалов, выбранных из группы связующих материалов, традиционных катализаторов гидропереработки, кислотных промоторов и крекирующих соединений, до, во время или после смешения и/или взаимодействия соединений металлов,
(v) распылительной сушки, (мгновенной) сушки, измельчения, замешивания, суспензионного смешения, сухого или мокрого смешения или их комбинации,
(vi) формования,
(vii) сушки и/или термической обработки при температуре ниже температуры, при которой происходит переход в кристаллическую структуру, предпочтительно ниже 450шС и
(viii) сульфидирования.
15. Способ гидропереработки углеводородного сырья, включающего серосодержащие и азотсодержащие органические соединения, включающий стадию контактирования углеводородного сырья с катализатором по п.1.
Текст
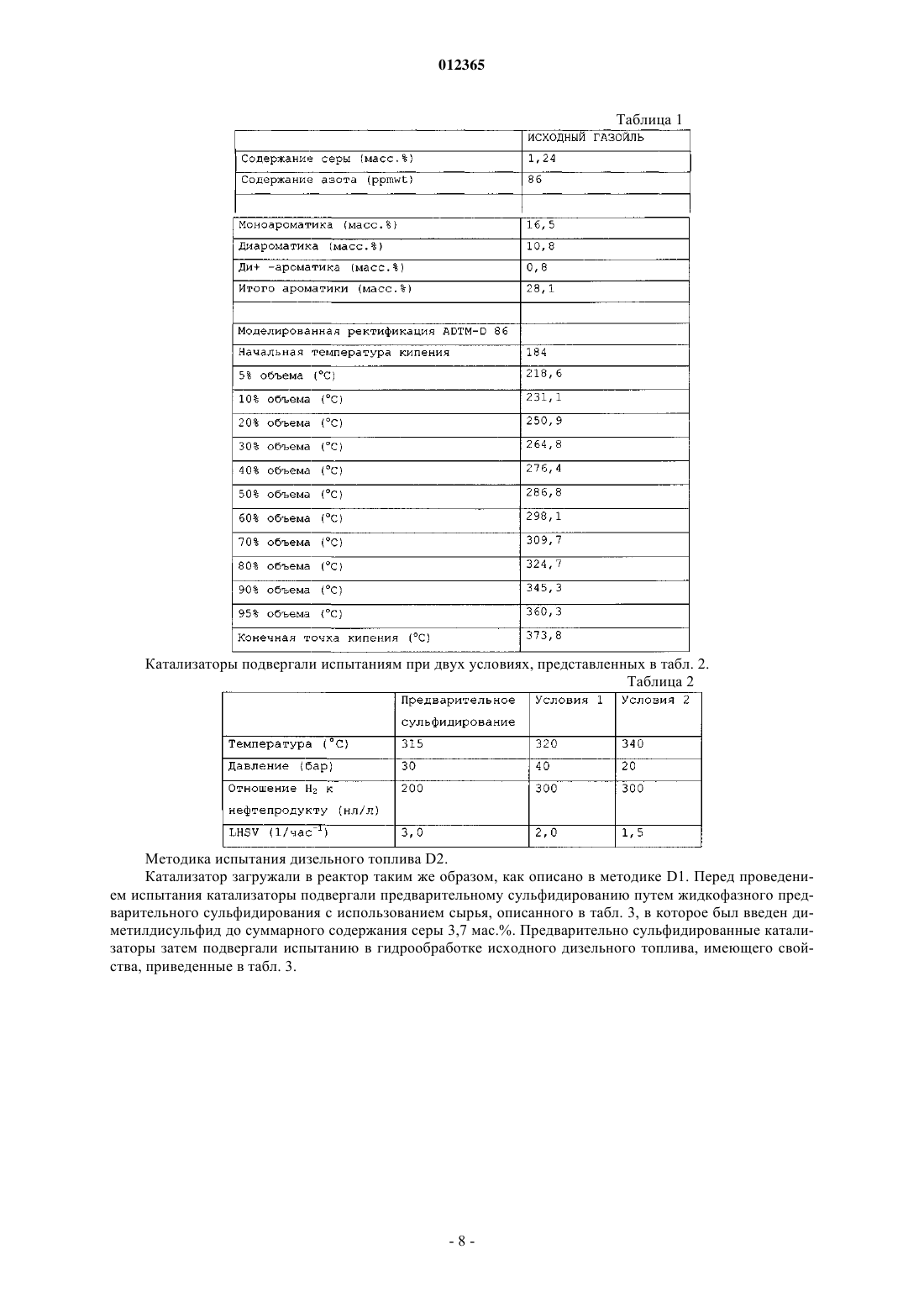
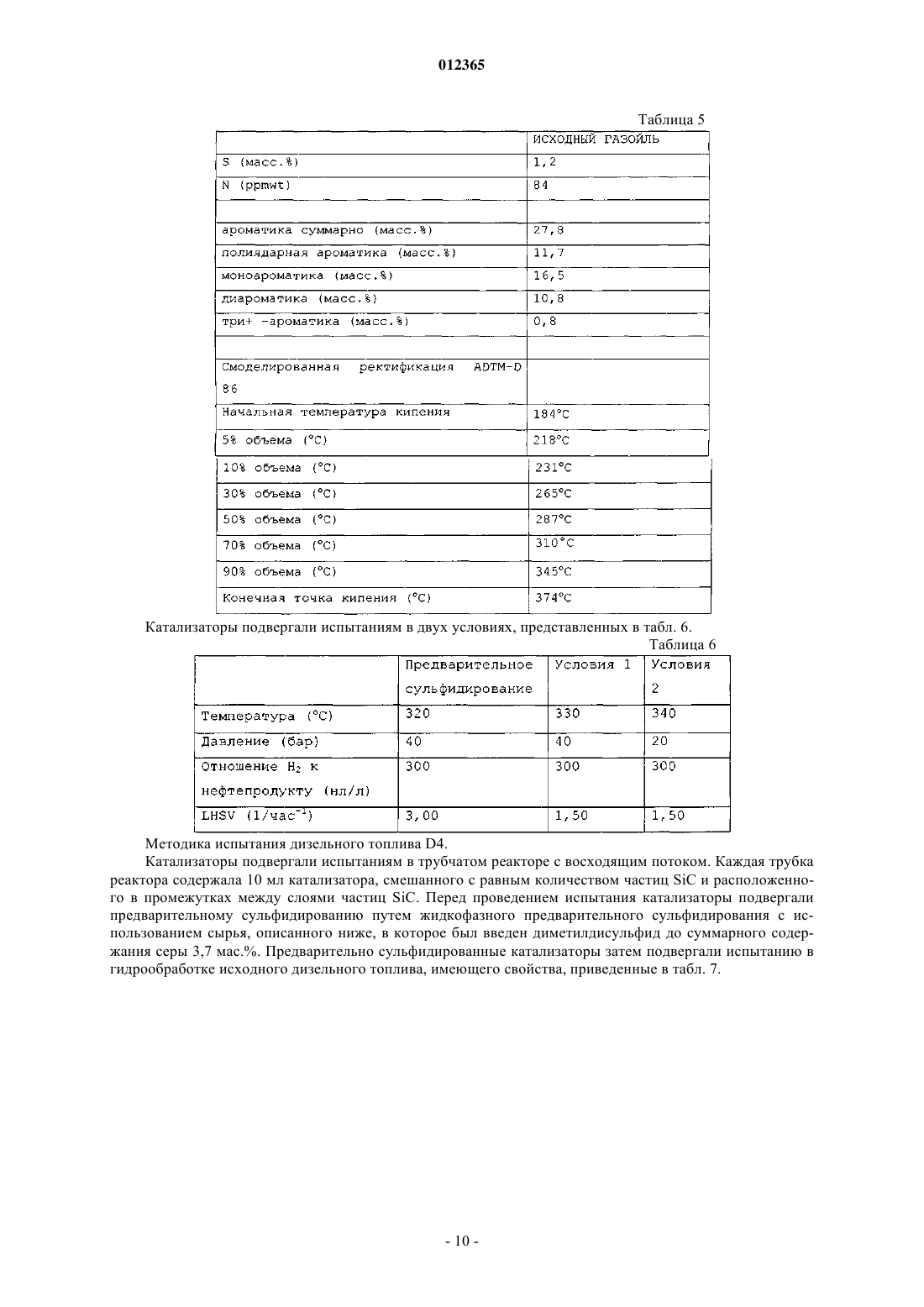
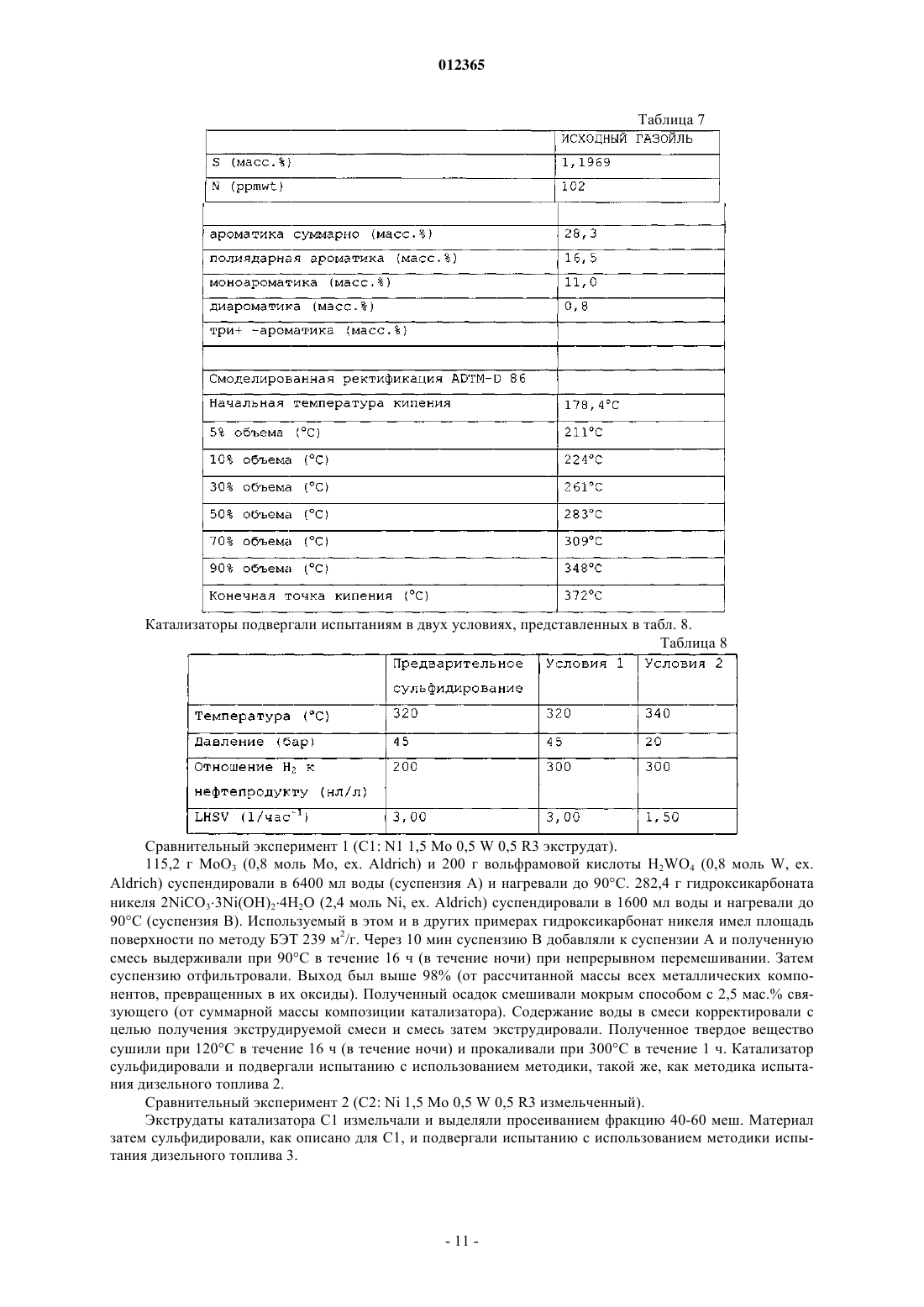
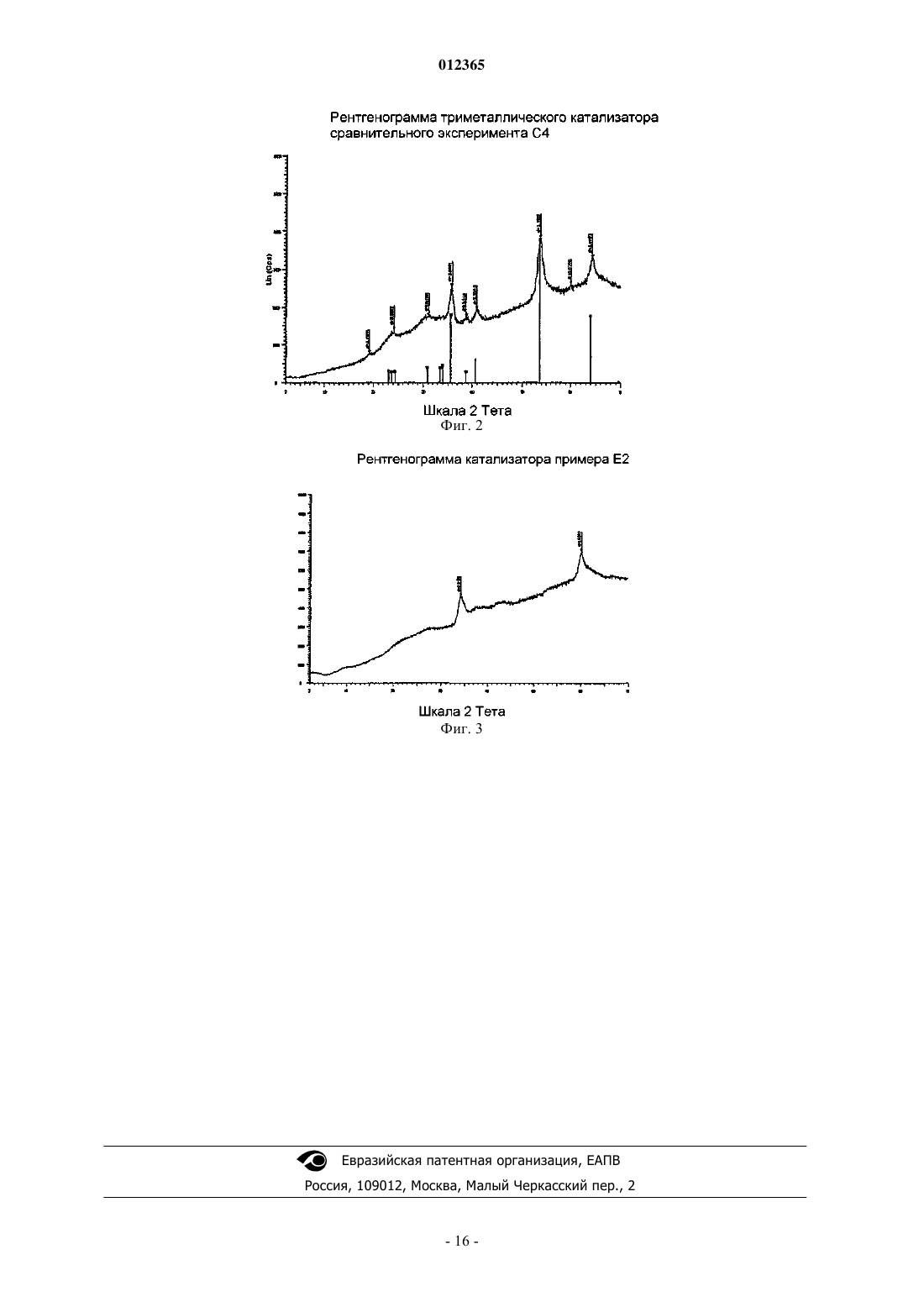
012365 Изобретение относится к насыпному катализатору, включающему по меньшей мере 60 мас.% частиц оксидов металлов, содержащих металл группы VIII и металл группы VIB молибден. Изобретение,кроме того, относится к способу получения насыпного катализатора, к соответствующему сульфидированному насыпному катализатору и к применению указанного или сульфидированного катализатора для гидропереработки, в частности гидрообессеривания и гидродеазотирования углеводородного сырья. Насыпной катализатор согласно изобретению обычно находится в виде формованных частиц, например, полученных экструзией композиции, включающей частицы оксидов металлов, и 0-40 мас.% (от суммарной массы насыпного катализатора) дополнительного материала, в частности связующего. Насыпной катализатор отличается от нанесенного на носитель катализатора тем, что он не включает предварительно отформованный материал носителя. Оксиды металлов не наносят на предварительно отформованный материал носителя, а они присутствуют в виде частиц оксидов металлов в формованном катализаторе. Насыпной катализатор, кроме того, отличается от нанесенных на носитель катализаторов тем,что насыпной катализатор включает по меньшей мере 60 мас.% частиц оксидов металлов (мас.%, вычисленных как отношение массы оксидов металлов к суммарной массе насыпного катализатора), в то время как нанесенные на носитель катализаторы содержат нанесенные на материал носителя оксиды металлов в количествах, значительно меньших чем 60 мас.%. Наиболее предпочтительно, чтобы насыпной катализатор являлся биметаллическим, т.е. содержащим практически только молибден в качестве металла группы VIB. Более подробно композиция насыпного катализатора описана ниже. Термин "гидропереработка или гидрообработка" в этом описании обычно охватывает все способы,при осуществлении которых углеводородное сырье взаимодействует с водородом при повышенной температуре и повышенном давлении, включая такие способы, как гидрирование, гидрообессеривание, гидродеазотирование, гидродеметаллизация, гидродеароматизация, гидроизомеризация, гидродепарафинизация, гидрокрекинг или гидрокрекинг в условиях умеренного давления, который обычно относят к мягкому гидрокрекингу. Далее, когда речь идет о высокой активности насыпного катализатора согласно изобретению, в частности, подразумевается активность при гидродеазотировании, если не указано иначе. Нанесенные на носитель биметаллические катализаторы и их применение для гидрообработки углеводородного сырья давно известны в технике. В патентном документе GB 820536 описан способ получения механически прочных частиц нанесенного на носитель катализатора, включающего комбинации кобальта, никеля, молибдена, ванадия или вольфрама, в котором используют высушенный при помощи распылительной сушки микросферический материал носителя из гидратированного оксида алюминия в количестве от 60 до 99 мас.% от суммарной массы катализатора. Катализаторы прокаливают при высокой температуре, например при 566 С в примере 1. В технике известны биметаллические никель-молибденовые насыпные катализаторы. Однако их характеризуют как уступающие по свойствам триметаллическим насыпным катализаторам, которые включают два металла вместо только одного металла группы VIB. В патентном документе WO 00/41810 описаны триметаллические насыпные катализаторы, включающие частицы насыпного катализатора, содержащие по меньшей мере один металл группы VIII и по меньшей мере два металла группы VIB, в частности катализаторы на основе никеля/молибдена/вольфрама. Частицы триметаллического насыпного катализатора получают с помощью способа, в котором соединения металлов смешивают в присутствии протонной жидкости и в котором по меньшей мере одно из соединений металлов остается, по меньшей мере частично, в твердом состоянии в течение всего времени осуществления способа. В сравнительном примере А описан никельмолибденовый насыпной катализатор, получаемый взаимодействием одного твердого соединения, включающего металл группы VIII, и одного растворенного соединения, включающего металл группы VIB. Полученные частицы оксидов металлов прокаливали при 400 С. Получаемые триметаллические насыпные катализаторы имеют значительно более высокую каталитическую активность, чем биметаллический насыпной катализатор, описываемый в сравнительных примерах А и В. В патентном документе WO 00/41811 описаны триметаллические насыпные катализаторы для гидропереработки и способ их получения, включающий стадии смешения и взаимодействия, по меньшей мере, соединения одного металла группы VIII в растворе, по меньшей мере, с соединениями двух металлов группы VIB в растворе в реакционной смеси с получением осадка. Получаемые частицы оксидов металлов прокаливали при 400 С. В сравнительном примере 2 описан биметаллический насыпной катализатор (кобальт/молибден). Получаемые триметаллические насыпные катализаторы имеют значительно более высокую каталитическую активность, чем биметаллический насыпной катализатор. В патентном документе ЕР 2005/004265 (предварительно неопубликованном) описаны триметаллические насыпные катализаторы для гидропереработки, включающие металл группы VIII, в частности никель, кобальт, железо или их смеси, металл группы V1B, в частности молибден, вольфрам или их смеси, и металл группы V в заданном молярном отношении. Молярное отношение металлов группы VIB к металлам группы V обычно составляет от 0,1 до 1, предпочтительно от 0,3 до 3.-1 012365 В патентном документе WO 99/03578 описан триметаллический насыпной катализатор для гидрообработки, в котором по меньшей мере часть, но не весь молибден в никель-молибденовом катализаторе замещен вольфрамом. Катализатор получают разложением (разложением при кипячении) предшественника из молибдовольфрамата никеля-аммония из раствора, или непосредственным осаждением растворенных солей металлов из раствора. В сравнительных примерах описан биметаллический никельмолибденовый насыпной катализатор (NH4Ni1Mo1-O), который получали разложением в результате кипячения раствора аммонийного комплекса металла. Получаемые частицы оксидов металлов прокаливали при 400 С. Получаемые триметаллические насыпные катализаторы имеют значительно более высокую каталитическую активность, чем биметаллический насыпной катализатор. В патентном документе WO 2004/073859 описан способ получения насыпного катализатора из оксида металла, включающего один или более металлов группы VIII и один или более металлов группыVIB в форме их оксидов или сульфидов, и тугоплавкий оксид. Способ включает регулируемое осаждение соединений металлов, материала из тугоплавкого оксида и щелочного соединения (предпочтительно аммонийсодержащих соединений) в протонной жидкости, с образованием аммонийного комплекса металла и материалов из тугоплавких оксидов, который затем нагревают. В примерах раскрыты биметаллические никель-молибденовые катализаторы. Заявлено, что способ прототипа приводит к насыпным катализаторам, которые практически аморфны и отличаются тем, что в их рентгенограмме отсутствует отражение,имеющее полную ширину на половине максимума 2,5 или менее. В патентном документе WO 2005/005582 описан способ получения смазывающего базового масла, в котором применяется насыпной катализатор для гидропереработки, включающий один или более металлов группы VIII и один или более металлов группы VIB в форме их оксидов или сульфидов, и тугоплавкий оксид. Описанными в примерах насыпными катализаторами являются биметаллические, в частности никель-молибденовый и никель-вольфрамовый, и их получают взаимодействием одного твердого соединения, включающего металл группы VIII, и одного растворенного соединения, включающего металл группы VIB, в присутствии тугоплавкого металла после добавления раствора аммония. Указывается, что структура получаемых частиц оксидов, определенная с помощью рентгеновского анализа, была аморфной. Приведенные выше ссылки на уже известный уровень техники позволяет сделать вывод, что триметаллические насыпные катализаторы имеют более высокую активность при гидрообессеривании по сравнению с биметаллическими насыпными катализаторами. Однако триметаллические катализаторы имеют недостаток по сравнению с биметаллическими катализаторами из-за того, что в силу присутствия в них двух различных соединений металлов группы VIB процесс получения катализаторов является более сложным. Дополнительным особым недостатком триметаллических катализаторов является трудность регенерации металлов из отработанных (или использованных) катализаторов, так как трудно разделить два различных металла группы VIB с высоким выходом. Главной задачей изобретения является разработка катализатора, который имеет высокую активность в способах гидрообессеривания и гидродеазотирования, применение которого позволяет достигать очень низких концентраций остаточной серы и азота в подвергнутом обработке сырье, который относительно просто получать, и в случае которого относительно просто регенерировать составляющие его металлы для повторного использования. Согласно изобретению предлагается насыпной катализатор, включающий по меньшей мере 60 мас.% частиц оксидов металлов, содержащих один или более металлов группы VIII и металл группыVIB молибден, содержащих менее чем 10 мол.% второго металла группы VIB (от суммарного количества металлов группы VIB) и содержащих металл группы V в количестве менее чем 10 мол.% (от суммарного количества металлов группы VIB), где насыпной катализатор прокален при температуре ниже температуры, при которой гексагональная метастабильная кристаллическая структура превращается в неактивную кристаллическую структуру, предпочтительно ниже 450 С, где насыпной катализатор имеет метастабильную гексагональную фазу, характеризуемую рентгенограммой, имеющей отражение при 33-35 и 58-61 2. Эту фазу называют метастабильной гексагональной фазой даже в том случае, если структура фактически может слегка отклоняться от строгой гексагональной структуры. Эти отражения сравнительно четко разрешаются. Было неожиданно обнаружено, что катализатор согласно изобретению имеет, в частности, высокую активность при гидродеазотировании дизельного топлива (далее хороший и лучший катализатор в этом тексте описания, в частности, означает насыпной катализатор с высокой или более высокой активностью при гидродеазотировании). Катализатор согласно изобретению позволяет достигать очень низких концентраций остаточного азота. Было обнаружено, что активность при гидродеазотировании, в частности,выраженная на единицу массы, является очень высокой по сравнению с активностью триметаллического насыпного катализатора известного уровня техники. Даже при сравнении на основе удельной объемной активности (или содержания остаточного азота) активность является все еще хорошей. Насыпной катализатор согласно изобретению особенно подходит для применения в комбинации с катализатором гидрообессеривания.-2 012365 Принимая во внимание сущность прототипа WO 2004/073859, было неожиданно обнаружено, что такую высокую активность можно было бы обнаружить в катализаторе, который не является аморфным,а, наоборот, имеет четко выраженные кристаллографические характеристики. Насыпной катализатор согласно изобретению имеет метастабильную гексагональную структуру, характеризующуюся рентгенограммой, показывающей два сравнительно четко разделяемых отражения: одно между 33 и 35, другое между 58 и 61 и главные отражения предпочтительно имеют полную ширину на половине максимума(FWHM) менее чем 2,5. Считается, что, кроме метастабильной гексагональной фазы, в катализаторе согласно изобретению могут присутствовать также некоторые аморфные фазы. Однако очевидно, что, так как присутствие метастабильной гексагональной фазы является индикатором высокой каталитической активности, предпочтительно в противоположность аморфному катализатору, описанному в прототипе WO 2004/073859,чтобы насыпной катализатор согласно изобретению характеризовался рентгенограммой, на которой главные отражения имеют полную ширину на половине максимума (FWHM) менее чем 2,5. Часто наблюдаются FWHM менее чем 2,0 или даже менее чем 1,5. В патентном документе US 2005/0065384 описан способ гидрирования оксоальдегидов. Катализатором в этом способе является восстановленным никель-молибденовым насыпным катализатором в противоположность настоящему изобретению, в котором используется оксидный насыпной катализатор. В этом документе также описано оксидное промежуточное соединение указанного восстановленного катализатора. Однако катализатор согласно изобретению является новым по отношению к этому катализатору прототипа в силу того, что его прокалили при температуре ниже температуры, при которой гексагональная метастабильная кристаллическая структура превращается в неактивную кристаллическую структуру, предпочтительно ниже 400 С. Кроме того, в противоположность прототипу катализатор согласно изобретению является катализатором гидропереработки, который перед использованием в способе гидропереработки предварительно сульфидируют in situ или ex-situ, предпочтительно предварительно сульфидируют в жидкой фазе, предпочтительно с помощью углеводородного сырья, содержащего вводимые в него сульфидирующие вещества. Было обнаружено, что частицы оксидов металлов в насыпном катализаторе согласно изобретению являются особенно чувствительными к термической обработке. Важно, чтобы насыпной катализатор, в частности частицы оксидов металлов в насыпном катализаторе, был подвергнут термической обработке при температуре ниже температуры, при которой происходит переход к кристаллической структуре. Это применимо к любой и ко всем стадиям термической обработки в способе получения насыпного катализатора, в частности при сушке и прокаливаниях частиц оксидов металлов или формованных частиц насыпного катализатора после смешения и формования. Предпочтительно, чтобы, насыпной катализатор подвергали термической обработке ниже 450 С, более предпочтительно ниже 400 С, еще более предпочтительно ниже 375 С и наиболее предпочтительно ниже 350 С. Когда в этом тексте описания имеется в виду "переход к кристаллической структуре", подразумевается кристаллическая структура, отличная от метастабильной гексагональной фазы. Считается, что неактивной высокотемпературной кристаллической структурой является бета-NiMoO4 или альфа-NiMoO4. Насыпной катализатор согласно изобретению в основном включает только молибден в качестве металла группы VIB. Насыпной катализатор может необязательно дополнительно включать второй металл группы VIII, например никель и кобальт, но наиболее предпочтительно, чтобы он также содержал только один металл группы VIII, предпочтительно никель. Насыпной катализатор может необязательно дополнительно включать менее чем 10 мол.% второго металла группы VIB (от суммарного количества металлов группы VIB). Наиболее предпочтительно, чтобы насыпной катализатор в основном включал только никель и молибден. Биметаллический насыпной катализатор может отличаться от триметаллического катализатора известного уровня техники тем, что он включает менее чем 10 мол.% второго металла группы VIB (от суммарного количества металлов группы VIB), но предпочтительно включает практически только один металл группы VIB молибден. Термин "практически только один металл группы VIB или группы VIII" означает, что наиболее предпочтительно, чтобы катализатор не содержал других металлов, но мог бы включать несущественное количество другого металла группы VIB или группы VIII, предпочтительно менее 5, более предпочтительно менее 3 и наиболее предпочтительно менее 1 мол.% (от суммарного количества металлов группыVIB или группы VIII). Насыпной катализатор может необязательно дополнительно включать менее чем 10 мол.% металла группы V (от суммарного количества металлов VIB). Это отличает его от предварительно неопубликованного патентного документа ЕР 2005/004265, описывающего насыпные катализаторы, включающиеNiMo насыпные катализаторы, содержащие металл группы V в количествах обычно от 10 до 90 мол.%(от суммы металлов группы VIB). В конкретном варианте осуществления насыпной катализатор согласно изобретению включает металл группы V, предпочтительно ниобий, в количестве от 0,1 до 10 мол.%(от суммы металлов группы VIB), предпочтительно от 0,1 до 9 мол.%, еще более предпочтительно от 0,1 до 7 мол.%. Хорошие результаты могут даже быть получены с металлом группы V в количестве от 0,1 до-3 012365 5 мол.%. Было обнаружено, что металл группы V повышает активность, даже когда присутствует в относительно малых количествах. В насыпном катализаторе согласно изобретению молярное отношение металла группы VIII к металлу группы VIB (далее называемое молярным отношением металлов), в частности молярное отношение никеля к молибдену, может в принципе изменяться в широких интервалах, например от 0,2 до 5. Обычно хорошие результаты могут быть получены при мольном отношении металлов от 0,2 до 4. Однако было обнаружено, что при низком отношении Ni/Mo, обычно от 0,2 до 1,5, очень часто не получали или полностью не получали гексагональную метастабильную структуру и/или FWHM (отражения метастабильной гексагональной фазы) составляла более чем 2,5. Часто обнаруживали, что образцы были истинно кристаллическими с большим числом максимумов пиков в различных положениях, вероятно,вследствие присутствия некоторых исходных материалов или других кристаллических структур неактивных соединений. Одно из неактивных соединений было идентифицировано как фаза, аналогичная фазе I, приведенной в публикации P. Ricol, Comptes Rendus, vol. 256, 1963, 3125-3127. Любая гексагональная фаза, если она присутствует, вряд ли могла быть обнаружена среди всех других пиков кристаллической структуры. Было также обнаружено, что в этом интервале мольного отношения металловNi/Mo активность была значительно ниже. Нижняя граница мольного отношения металлов может зависеть от используемых конкретных условий способа, но обычно предпочтительно, чтобы мольное отношение металла группы VIII к металлу группы VIB, в частности мольное отношение никеля к молибдену,составляло больше 1,5, более предпочтительно больше 2, еще более предпочтительно больше 2,5 и наиболее предпочтительно более 3. Следует отметить, что это наблюдение также отличается от сущности прототипа WO 2004/073859, в котором описано, что мольное отношение металлов Ni/Mo обычно составляет от 1 до 2 и наиболее предпочтительно 1. Кроме того, было обнаружено, что при низком мольном отношении металлов сложно, если не невозможно, получить хороший катализатор при описанных выше стандартных атмосферных условиях проведения реакции, но можно получить хороший катализатор с помощью гидротермального способа, в котором реакцию осуществляют при гидротермальных условиях при температуре реакции выше, чем температура кипения протонной жидкости при атмосферном давлении. При гидротермальных условиях предпочтительно, чтобы протонной жидкостью являлась вода и реакцию осуществляли при гидротермальных условиях при температуре реакции выше 100 С, предпочтительно при давлении реакции выше чем 1 бар. Изобретение также относится к насыпному катализатору согласно изобретению, имеющему молярное отношение никеля к молибдену от 0,2 до 5, получаемому по этому гидротермальному способу. По многим причинам вода является самым хорошим выбором в качестве протонной жидкости. Однако не исключаются и другие протонные жидкости и поэтому предполагается, что "гидротермальные условия реакции" в этом тексте описания также охватывают реакционные условия, в которых используют протонную жидкость, отличную от воды, давления выше атмосферного давления и температуры выше температуры кипения протонной жидкости. Предпочтительно, чтобы температура реакции составляла по меньшей мере на 10%, более предпочтительно по меньшей мере на 25%, еще более предпочтительно по меньшей мере на 50% и наиболее предпочтительно по меньшей мере на 75% выше температуры кипения протонной жидкости. Предпочтительно, чтобы реакция осуществлялась в воде в качестве протонной жидкости при температуре реакции по меньшей мере 110, предпочтительно по меньшей мере 125, еще более предпочтительно по меньшей мере 150 и наиболее предпочтительно по меньшей мере 175 С предпочтительно в автоклаве, предпочтительно при соответствующем повышенном давлении. Хорошие результаты могут быть получены при температурах реакции от 110 до 170 С. В предпочтительном варианте осуществления изобретения реакционную смесь нагревают с помощью микроволнового излучения. Растворенные компоненты в реакционной смеси могут повышать температуру кипения протонной жидкости. Предпочтительно с точки зрения достижения высокой активности, чтобы в этих гидротермальных условиях мольное отношение металла группы VIII к металлу группы VIB составляло больше чем 0,3, предпочтительно больше чем 0,4, более предпочтительно больше чем 0,5, еще более предпочтительно больше чем 0,6 и наиболее предпочтительно больше чем 0,7. Насыпной катализатор включает по меньшей мере 60 мас.% частиц оксидов металлов (мас.%, вычисленные в пересчете на оксиды металлов от суммарной массы насыпного катализатора), чем он отличается от нанесенных на носитель катализаторов, которые содержат оксиды металлов, нанесенные на материал носителя в количествах, значительно меньших чем 60 мас.%. Предпочтительно, чтобы насыпной катализатор согласно изобретению включал по меньшей мере 70 мас.%, более предпочтительно по меньшей мере 75 мас.%, еще более предпочтительно по меньшей мере 80 мас.% и наиболее предпочтительно по меньшей мере 85 мас.% частиц оксидов металлов, при этом предпочтительно, чтобы оставшиеся от 0 до 40 мас.% являлись одним или более материалами, выбранными из группы связующих материалов, традиционных катализаторов гидропереработки, кислотных промоторов и крекирующего компонента. Обычно после смешения частиц оксидов металлов со связующим композицию формуют, предпочтительно экструдируют, с получением формованных частиц насыпного катализатора. Изобретение также относится к формованным частицам насыпного катализатора,-4 012365 включающим частицы оксидов металлов. В качестве варианта частицы оксидов металлов насыпного катализатора могут быть использованы непосредственно в способе гидропереработки, т.е. практически без смешения и формования, например распылительной сушкой. Это называют способом суспензионной гидрообработки. Для этого использования предпочтительно, чтобы частицы подвергали обработке с получением частиц с узким распределением по размеру, например путем просеивания или агломерации, но практически без смешения и формования. Изобретение также относится к использованию металлического катализатора согласно изобретению в суспензионном способе гидрообработки, предпочтительно с использованием насыпного катализатора из частиц оксидов металлов практически без смешения и формования. Стадия i). На первой стадии способа получения насыпного катализатора согласно изобретению реагирующие соединения смешивают с образованием реакционной смеси. Это может быть осуществлено множеством различных способов, например, описанных в патентных документах WO 00/41810, WO 99/03578,WO 2004/073859, WO 2005/005582 и WO 00/41811. Первое и/или второе соединения могут быть растворимыми или, по меньшей мере, частично растворимыми в протонной жидкости. Можно сначала получать суспензию или раствор соединения металла в протонной жидкости и добавлять, одновременно или один за другим, раствор (растворы) и/или дополнительно суспензию (суспензии), включающие растворенное и/или суспендированное соединение (соединения) металла в протонной жидкости. Также возможно сначала смешивать растворы или одновременно, или один за другим и последовательно добавлять дополнительную суспензию (суспензии) и необязательно раствор (растворы) или одновременно, или один за другим. Однако в предпочтительном варианте осуществления способ получения насыпного катализатора согласно изобретению включает в себя i) получение реакционной смеси соединения первого металла,включающего металл группы VIII, и соединения второго металла, включающего металл группы VIB молибден, необязательно включающей одно или более дополнительных соединений, включающих второй металл группы VIB в количестве менее чем 10 мол.% (от суммарного количества металлов группы VIB) и/или металла группы V в количестве менее чем 10 мол.% (от суммы металлов группы VIB) и/или второй металл группы VIII в протонной жидкости и ii) взаимодействие соединений первого и второго металлов при повышенной температуре, при котором твердое соединение первого и/или второго металла остается,по меньшей мере, частично в твердом состоянии в процессе всей реакции с образованием частиц оксидов металлов, где насыпной катализатор на всех стадиях в процессе его получения остается при температуре ниже температуры, при которой гексагональная метастабильная кристаллическая структура превращается в неактивную кристаллическую структуру, предпочтительно ниже 400 С. Предпочтительно, чтобы в этом способе соединение первого металла и соединение второго металла оставалось, по меньшей мере, частично в твердом состоянии в процессе всей реакции. Этот предпочтительный способ называют способом "твердое вещество - твердое вещество". Этот способ является относительно простым, характеризуется высоким выходом и не представляет опасности для окружающей среды, но наиболее важным является то, что получаемый указанным способом насыпной катализатор является высокоактивным. Термин "по меньшей мере, частично в твердом состоянии" означает, что по меньшей мере часть соединения металла присутствует в виде твердого соединения металла и, необязательно, другая часть соединения металла присутствует в виде раствора этого соединения металла в протонной жидкости. Этот способ "твердое вещество - твердое вещество" описан подробно в патентном документе WO 00/41810 в качестве одного из нескольких возможных путей получения триметаллического насыпного катализатора. Предпочтительно с точки зрения достижения высокого выхода и низкого неблагоприятного воздействия на окружающую среду, чтобы соединения первого и второго металла не содержали атомов азота и чтобы протонную жидкость, отделенную от прореагировавших частиц оксидов металлов, повторно использовали, по меньшей мере, частично для образования суспензии на стадии i). Наиболее предпочтительно, чтобы в этом способе соединением первого металла являлся (гидроксикарбонат) карбонат металла и соединением второго металла являлся оксид металла или кислота. С точки зрения получения высокоактивного катализатора, кроме того, предпочтительно, чтобы в способе первым соединением являлся карбонат или гидроксикарбонат никеля, имеющий площадь поверхности по меньшей мере 150 м 2/г. По ряду причин этот предпочтительный способ является наиболее экологически безопасным и экономически оптимальным способом получения катализатора. Кроме того, что соединения металлов не содержат атомов азота, реакция также не требует добавления аммиака к реакционной смеси, как, например, в патентном документе WO 2004/073859, поэтому в способе полностью отсутствуют атомы азота. Способ характеризуется отсутствием накопления инородных ионов, таких как аммоний и/или нитрат, в протонной жидкости при повторяющихся операциях рецикла, отсутствием жесткой необходимости в промывке полученных отделенных оксидных частиц, меньшим неблагоприятным воздействием на окружающую среду вследствие снижения количества отходов тяжелых переходных металлов и отсутствием опасности взрывов вследствие образования соли нитрата аммония. Кроме того, так как катализатор является биметаллическим, химическое взаимодействие на стадии реакции является более простым, так как там имеет-5 012365 ся только один металл группы VIB и поэтому не происходит изменение состава металлов группы VIB в композиции при рецикле отделенной жидкости после реакции. Так как соединения остаются, по меньшей мере, частично твердыми в ходе всей реакции, количество металлов, растворенных в протонной жидкости, является небольшим и, следовательно, потери составляют меньшую величину. Кроме того, отработанный биметаллический катализатор более легко перерабатывать на составляющие металлы для повторного использования, чем триметаллический катализатор, так как нет необходимости в разделении двух металлов группы VIB, что является очень сложным. Могут быть использованы традиционные способы разделения никеля и молибдена. Это является преимуществом с точки зрения снижения сложности способа повторного использования, снижения затрат и повышения степени извлечения металла. В альтернативном варианте осуществления получение насыпного катализатора осуществляют способом, включающим i) образование раствора растворимого соединения первого металла, содержащего металл группы VIII, и растворимого соединения второго металла группы VIB молибдена, при этом раствор необязательно дополнительно включает дополнительное соединение, включающее второй металл группы VIB в количестве меньше чем 10 мол.% (от суммарного количества металлов группы VIB), и необязательно соединение, включающее соединение металла группы V в количестве меньше чем 10 мол.%(от суммы металлов группы VIB), и необязательно второе соединение металла группы VIII, и ii) взаимодействие и/или осаждение соединений первого и второго металлов с образованием частиц оксидов металлов, где насыпной катализатор на всех стадиях в процессе его получения остается при температуре ниже температуры, при которой гексагональная метастабильная кристаллическая структура превращается в неактивную кристаллическую структуру, предпочтительно ниже 400 С. Для практически полного завершения реакции время реакции выбирают достаточно большим. Реакцию завершают тогда, когда на рентгенограмме отделенных частиц оксидов металлов перестают присутствовать отражения непрореагировавших исходных соединений. В любом случае время реакции выбирают так, чтобы на рентгенограмме готового насыпного катализатора после сушки, формования и прокаливания отсутствовали отражения непрореагировавших исходных веществ. В способе получения, в котором по меньшей мере одно или все из участвующих в реакции соединений находятся, по меньшей мере, частично в твердом состоянии в процессе всей реакции, реакцию обычно проводят в течение по меньшей мере 2 ч, предпочтительно по меньшей мере 4 ч, более предпочтительно по меньшей мере 6 ч и наиболее предпочтительно по меньшей мере 8 ч. Особым преимуществом способа гидротермальной реакции является более высокая скорость реакции соединений металлов или, наоборот, в принципе более высокий выход в течение одного и того же времени реакции. Это является преимуществом, особенно когда необходимо низкое мольное отношение металла группы VIII к металлу группы VI, так как было обнаружено, что низкое мольное отношение металлов снижает скорость реакции во время получения. Обычно стадия суспендирования и смешения исходных материалов не является определяющей при условии, что для обеспечения хорошего перемешивания суспензии присутствует достаточное количество растворителя. Кроме того, в случае очень реакционно-способных и/или частично растворимых исходных материалов следует предотвращать бурное взаимодействие исходных материалов уже во время их добавления. Это может быть опять достигнуто путем, например, увеличения количества растворителя или понижения температуры, при которой смешивают материалы. У любого специалиста в этой области не возникнет проблем при выборе подходящих условий. Способ необязательно дополнительно включает одну или более из следующих обычных стадий: iii) отделения частиц оксидов металла от реакционной смеси; iv) смешения частиц оксидов металлов от 0 до 40 мас.% одного или более материалов, выбранных из группы связующих материалов, традиционных катализаторов гидропереработки, кислотных промоторов и крекирующих соединений до, во время или после смешения и/или взаимодействия соединений металлов; v) распылительной сушки, (мгновенной) сушки, измельчения, замешивания, суспензионного смешения, сухого или мокрого смешения или их комбинаций; vi) формования; vii) сушки и/или термической обработки при температуре ниже температуры, при которой происходит переход к кристаллической структуре, предпочтительно ниже 400 С и vii) сульфидирования. Изобретение, кроме того, относится к насыпному катализатору, получаемому описанным выше способом согласно изобретению; к сульфидированному насыпному катализатору, включающему насыпной катализатор согласно изобретению; к применению насыпного катализатора или сульфидированного насыпного катализатора для гидропереработки углеводородного сырья, включающего серосодержащие и азотсодержащие органические соединения; и к способу исключительно глубокого гидрообессеривания серосодержащего и азотсодержащего углеводородного сырья, включающему контактирование сырья с(несульфидированным) сульфидированным насыпным катализатором согласно изобретению. Насыпной катализатор согласно изобретению особенно подходит для применения в комбинации с очень хорошим катализатором гидрообессеривания. Высокая активность при гидродеазотировании и чрезвычайно низкие концентрации остаточного азота позволяют при использовании катализатора для гидрообессеривания достигать низких концентраций остаточной серы. Содержание остаточного азота, т.е. количество азота (в ppmwt) после гидрообработки с использованием катализатора, составляет ниже 10, предпочти-6 012365 тельно ниже 5, более предпочтительно ниже 2, еще более предпочтительно ниже 1 ppmwt. Это особенно применимо к дизельному топливу, в котором исходное содержание N является низким, но также и к предварительной обработке сырья для установки гидрокрекинга, где исходное содержание N часто является высоким, и к гидропереработке в условиях высокого давления, например при давлении выше 20 бар,более предпочтительно выше 30 бар и наиболее предпочтительно выше 40 бар. Насыпной катализатор изобретения особенно подходит для гидрообработки углеводородного сырья. Такие способы гидрообработки включают, например, гидрообессеривание, гидродеазотирование и гидродеароматизацию углеводородного сырья. Подходящим сырьем являются, например, газойль, керосин, нафта, вакуумные газойли и тяжелые газойли. Могут быть использованы традиционные условия осуществления способа, такие как температуры в интервале 250-450 С, давления в интервале 5-250 бар,объемные скорости в интервале 0,1-10 ч-1 и отношения Н 2/нефтепродукт в интервале 50-2000 нл/л. Насыпной катализатор согласно изобретению может быть использован фактически во всех способах гидропереработки для обработки множества видов сырья при широком интервале реакционных условий, например при температурах в интервале от 200 до 450 С, давлениях водорода от 5 до 300 бар и объемных скоростях (LHSV) в интервале от 0,05 до 10 ч-1. Характеристическая полная ширина на половине максимума. Характеристическая полная ширина на половине максимума FWHM оксидных катализаторов определяли на основе рентгенограммы катализатора: FWHM является полной шириной на половине максимума (в единицах 2 угла рассеивания отражения от 33 до 35 и угла рассеивания от 58 до 61. Для получения рентгенограммы может быть использован стандартный порошковый дифрактометр, оборудованный графитовым монохроматором. Могут быть выбраны следующие условия измерения, например, установки генератора рентгеновского излучения: 40 кВ и 40 мА, длина волны: 1,5418 ангстрем, щель расхождения луча и антирассеивающая щель: v20 (переменная), щель детектора: 0,6 мм, размер шага: 0,05 (2), время/шаг: 2 с, прибор: Bruker D5000. Для определения максимума отражений проводят известную для специалиста корректировку измеренной рентгенограммы с учетом нулевой линии и/или с учетом фонового рассеяния. Изобретение будет дополнительно проиллюстрировано приведенными ниже примерами.R3 означает способ осуществления реакции, в котором и соединение первого металла, и соединение второго металла являются, по меньшей мере, частично твердыми в процессе реакции.R2 означает способ осуществления реакции, в котором по меньшей мере одно из соединений первого металла или второго металла является, по меньшей мере, частично твердым в процессе реакции, а другие соединения являются растворенными.CBD означает насыпную плотность компактного катализатора. Результаты испытания по гидропереработке дизельного топлива приведены в табл. 9, где RVA и RWA являются относительной объемной активностью и относительной массовой активностью соответственно в расчете на суммарное количество катализатора, загруженного в реактор.HDN обозначает гидродеазотирование и HDS обозначает гидрообессеривание. Испытания проводили с использованием двух условий испытаний 1 и 2 при различных температурах и давлении. Постфиксное обозначение 1 или 2 (например, в RWA1 и RWA2) относится к условиям испытаний 1 и 2 соответственно. При испытаниях были использованы различные методики испытаний дизельного топлива, обозначаемые как D1, D2, D3 и D4. Значения RWA/RVA для катализаторов сравнения С 3, С 1, С 2 в методиках испытаний дизельного топлива D1, D2, D3 соответственно были заданы равными 100. Все другие RWA/RVA значения вычисляли относительно этих катализаторов сравнения. Эти условия испытаний и методика испытания дизельного топлива описаны более подробно ниже. Результаты для RWA HDN1 не приводятся, так как содержания азота в продукте реакции были все настолько низкими, что измерение является неточным и разницы между образцами являются слишком малыми для выявления разницы в каталитической активности между образцами. Кроме того, определяли содержание остаточного азота и серы после гидрообработки, которые приведены в табл. 9 в столбцах S1, S2 и N2. Методика испытания дизельного топлива D1. Катализаторы подвергали испытаниям в трубчатом реакторе с нисходящим потоком. Каждая трубка реактора содержала 10 мл катализатора, смешанного с равным количеством частиц SiC и расположенного в промежутках между слоями частиц SiC. Перед проведением испытания катализаторы подвергали предварительному сульфидированию путем жидкофазного предварительного сульфидирования с использованием сырья, описанного в табл. 1, в которое был введен диметилдисульфид до суммарного содержания серы 3,7 мас.%. Предварительно сульфидированные катализаторы затем подвергали испытанию при гидрообработке исходного дизельного топлива, имеющего свойства, приведенные в табл. 1. Катализаторы подвергали испытаниям при двух условиях, представленных в табл. 2. Таблица 2 Методика испытания дизельного топлива D2. Катализатор загружали в реактор таким же образом, как описано в методике D1. Перед проведением испытания катализаторы подвергали предварительному сульфидированию путем жидкофазного предварительного сульфидирования с использованием сырья, описанного в табл. 3, в которое был введен диметилдисульфид до суммарного содержания серы 3,7 мас.%. Предварительно сульфидированные катализаторы затем подвергали испытанию в гидрообработке исходного дизельного топлива, имеющего свойства, приведенные в табл. 3. Катализаторы подвергали испытаниям в двух условиях, представленных в табл. 4. Таблица 4 Методика испытания дизельного топлива D3. Катализатор загружали в реактор таким же образом, как описано в методике D1. Перед проведением испытания катализаторы подвергали предварительному сульфидированию путем жидкофазного предварительного сульфидирования с использованием сырья, описанного в табл. 5, в которое был введен диметилдисульфид до суммарного содержания серы 3,7 мас.%. Предварительно сульфидированные катализаторы затем подвергали испытанию в гидрообработке исходного дизельного топлива, имеющего свойства, приведенные в табл. 5. Катализаторы подвергали испытаниям в двух условиях, представленных в табл. 6. Таблица 6 Методика испытания дизельного топлива D4. Катализаторы подвергали испытаниям в трубчатом реакторе с восходящим потоком. Каждая трубка реактора содержала 10 мл катализатора, смешанного с равным количеством частиц SiC и расположенного в промежутках между слоями частиц SiC. Перед проведением испытания катализаторы подвергали предварительному сульфидированию путем жидкофазного предварительного сульфидирования с использованием сырья, описанного ниже, в которое был введен диметилдисульфид до суммарного содержания серы 3,7 мас.%. Предварительно сульфидированные катализаторы затем подвергали испытанию в гидрообработке исходного дизельного топлива, имеющего свойства, приведенные в табл. 7. Катализаторы подвергали испытаниям в двух условиях, представленных в табл. 8. Таблица 8Aldrich) суспендировали в 6400 мл воды (суспензия А) и нагревали до 90 С. 282,4 г гидроксикарбоната никеля 2NiCO33Ni(ОН)24 Н 2 О (2,4 моль Ni, ex. Aldrich) суспендировали в 1600 мл воды и нагревали до 90 С (суспензия В). Используемый в этом и в других примерах гидроксикарбонат никеля имел площадь поверхности по методу БЭТ 239 м 2/г. Через 10 мин суспензию В добавляли к суспензии А и полученную смесь выдерживали при 90 С в течение 16 ч (в течение ночи) при непрерывном перемешивании. Затем суспензию отфильтровали. Выход был выше 98% (от рассчитанной массы всех металлических компонентов, превращенных в их оксиды). Полученный осадок смешивали мокрым способом с 2,5 мас.% связующего (от суммарной массы композиции катализатора). Содержание воды в смеси корректировали с целью получения экструдируемой смеси и смесь затем экструдировали. Полученное твердое вещество сушили при 120 С в течение 16 ч (в течение ночи) и прокаливали при 300 С в течение 1 ч. Катализатор сульфидировали и подвергали испытанию с использованием методики, такой же, как методика испытания дизельного топлива 2. Сравнительный эксперимент 2 (С 2: Ni 1,5 Мо 0,5 W 0,5 R3 измельченный). Экструдаты катализатора С 1 измельчали и выделяли просеиванием фракцию 40-60 меш. Материал затем сульфидировали, как описано для С 1, и подвергали испытанию с использованием методики испытания дизельного топлива 3.- 11012365 Сравнительный эксперимент 3 (Cl: Ni 1,5 Мо 0,5 W 0,5 R3 экструдат). Экструдаты катализатора С 1 сульфитизировали и подвергали испытанию с использованием методики испытания дизельного топлива 1. Пример 1 (Е 1: N1 1,5 Мо 1 R2 экструдат). 282,4 г гептамолибдата аммония (NH4)6Mo7O24H2O (1,6 моль Мо, ex. Aldrich) растворяли в 6400 мл воды, корректируя рН раствора до 5,2 при комнатной температуре. Раствор затем нагревали до 90 С(раствор А). 282,4 г гидроксикарбоната никеля 2NiCO33Ni(ОН)24 Н 2 О (2,4 моль Ni, ex. Aldrich) суспендировали в 1600 мл воды и эту суспензию нагревали до 90 С (суспензия В). Затем через 10 мин суспензию В добавляли к раствору А и полученную суспензию выдерживали при 90 С в течение 16 ч при непрерывном перемешивании. Выход составлял выше 85%. Полученный осадок экструдировали (с 10 мас.% связующего), сушили, прокаливали и сульфидировали, как описано для С 1, и подвергали испытанию с использованием методики испытания дизельного топлива 2. Пример 2 (Е 2: Ni 3 Mo 1 R3 экструдат). Катализатор получали так же, как описано в сравнительном эксперименте 1 (С 1), за исключением того, что использовали только один металл группы VIB. Катализатор готовили с использованием 230,4 г триоксида молибдена (1,6 моль Mo, ex. Aldrich) и 564,8 г гидроксикарбоната никеля 2NiCO33Ni(ОН)24 Н 2 О (4,8 моль Ni). Выход составлял около 98% в расчете на рассчитанную массу всех металлических компонентов, превращенных в их оксиды. Получаемый осадок экструдировали(с 10 мас.% связующего), сушили, прокаливали и сульфидировали, как описано для С 1, и подвергали испытанию с использованием методики испытания дизельного топлива 2. Пример 3 (Е 3: Ni 2 Mo 1 порошок R3). Катализатор получали так же, как описано в сравнительном эксперименте 1 (С 1), за исключением того, что использовали только один металл группы VIB и готовили меньшее количество катализатора. Катализатор готовили с использованием 28,8 г триоксида молибдена (0,2 моль Mo, ex. Aldrich) и 47,1 г гидроксикарбоната никеля 2NiCO33Ni(ОН)24 Н 2 О (4,8 моль Ni). Выход составлял около 98% в расчете на рассчитанную массу всех металлических компонентов, превращенных в их оксиды. Полученное твердое вещество сушили при 120 С в течение 16 ч (в течение ночи) и прокаливали при 300 С в течение 1 ч. Полученный материал гранулировали, гранулы измельчали и отделяли просеиванием фракцию 40-60 меш. Материал затем сульфидировали, как описано для С 1, и подвергали испытанию с использованием методики испытания дизельного топлива 3. Пример 4 (Е 4: Ni 3 Мо 1 порошок R3). Катализатор получали так же, как описано в сравнительном эксперименте 1 (С 1), за исключением того, что использовали только один металл группы VIB и готовили меньшее количество катализатора. Катализатор готовили с использованием 28,8 г триоксида молибдена (0,2 моль Mo, ex. Aldrich) и 70,6 г гидроксикарбоната никеля 2NiCO33Ni(OH)24H2O (0,6 моль Ni). Выход составлял около 98% в расчете на рассчитанную массу всех металлических компонентов, превращенных в их оксиды. Полученное твердое вещество сушили при 120 С в течение 16 ч (в течение ночи) и прокаливали при 300 С в течение 1 ч. Полученный материал гранулировали, гранулы измельчали и отделяли просеиванием фракцию 40-60 меш. Материал затем сульфидировали, как описано для С 1, и подвергали испытанию с использованием методики испытания дизельного топлива 3. Пример 5 (Е 5: Co 2 Mo 1 порошок R3). Приготавливали катализатор и подвергали его испытанию так же, как описано в Е 3, за исключением того, что вместо гидроксикарбоната никеля (0,4 моль Ni) использовали гидроксикарбонат кобальта(0,4 моль Со). Пример 6 (Е 6: Ni 1,5 Mo 1 R3 экструдат). Катализатор получали так же, как описано в сравнительном эксперименте 1 (С 1), за исключением того, что использовали только один металл группы VIB. Катализатор готовили с использованием 230,4 г триоксида молибдена (1,6 моль Mo ex. Aldrich) и 282,4 г гидроксикарбоната никеля 2NiCO33Ni(ОН)24 Н 2 О (2,4 моль Ni). Выход составлял около 98% в расчете на рассчитанную массу всех металлических компонентов, превращенных в их оксиды. Получаемый осадок экструдировали(с 2,5 мас.% связующего), сушили, прокаливали и сульфидировали, как описано для С 1, и подвергали испытанию с использованием методики испытания дизельного топлива 1. Измеряли полную ширину на половине максимума (FWHM) для всех примеров и было обнаружено,что она составляет менее 2,5 во всех примерах Е 1-Е 6. Результаты в табл. 9 показывают, что катализатор согласно изобретению позволяет достигать очень низких концентраций остаточного азота. Активность при гидродеазотировании, особенно выраженная на единицу массы, является высокой по сравнению с триметаллическим насыпным катализатором известного уровня техники. Даже при сравнении на основе удельной объемной активности активность по-прежнему является хорошей по сравнению с триметаллическим насыпным катализатором известного уровня техники. Очевидно, что активность повышается с повышением содержания металла группы VIII. Сравнение условий 1 и 2 показывает, что результаты гидродеазотирования при более высоких давлениях улучшаются в неожиданной степени. Активность при- 12012365 гидрообессеривании является относительно низкой, но приемлемой. Насыпной катализатор согласно изобретению особенно подходит для использования в комбинации с очень хорошим катализатором гидрообессеривания. Высокая активность при гидродеазотировании и чрезвычайно низкие концентрации остаточного азота позволяют при применении катализатора гидрообессеривания достигать значительно более низких концентраций остаточной серы. Сравнительный эксперимент С 4 (Ni 1 W 0,5 Mo 0,5 R3). 188 г гидроксикарбоната никеля 2NiCO33Ni(ОН)24 Н 2 О (1,6 моль Ni) суспендировали в 8000 мл воды и полученную суспензию нагревали до 60 С. Затем добавляли 115,2 г MoO3(0,8 моль Мо) и 200 г вольфрамовой кислоты H2WO4 (0,8 моль W), полученную суспензию нагревали до 95 С и выдерживали при этой температуре в течение приблизительно 24 ч при постоянном перемешивании. После чего суспензию отфильтровывали. Полученный осадок смешивали мокрым способом с 10 мас.% связующего (от суммарной массы композиции катализатора). Корректировали содержание воды в смеси для получения эксрудируемой смеси и затем смесь экструдировали. Полученное твердое вещество сушили при 120 С в течение 16 ч (в течение ночи) и прокаливали при 300 С в течение 1 ч. Экструдаты измельчали и отделяли просеиванием фракцию 40-60 меш. Материал затем сульфидировали и подвергали испытанию с использованием методики испытания дизельного топлива D4. Сравнительный эксперимент С 5. Был воспроизведен способ, описанный в примере 1 в патентном документе WO 2004/073859. Смешивали 12,8 г ADM (0,075 моль Мо) и 11,0 г NiCO3 (0,092 моль Ni) и добавляли к 112,5 г Н 2 О в автоклав объемом 225 мл. Температуру увеличивали до 80 С при повышенном давлении. Разбавляли 5,65 г аммиака (25 мас.% раствор) с помощью 37,5 г H2O. В этом растворе диспергировали 4,61 г Sipernat 2200. Автоклав открывали (значительное избыточное давление отсутствовало) и к смеси в автоклаве добавляли суспензию диоксида кремния. Автоклав закрывали, нагревали до 80 С и выдерживали при 80 С в течение 30 мин. Автоклав открывали (значительное избыточное давление отсутствовало) и выделяли твердые частицы с помощью распылительной сушки при условиях, гарантирующих то, что твердые частицы не подвергаются воздействию температур выше 300 С (реальная температура не превышала 180 С). Вследствие применения распылительной сушки выход продукта должен быть 100%. Это соответствует (рассчитанной) композиции 48,4 мас.% МоО 3, 30,9 мас.% NiO и 20,7 мас.% SiO2. Полученное твердое вещество гранулировали, гранулы измельчали и отделяли просеиванием фракцию 40-60 меш. Материал затем прокаливали при 300 С в течение 1 ч. Материал затем сульфидировали и подвергали испытанию с использованием методики испытания дизельного топлива D4. Рентгенограмма композиции сравнительного эксперимента С 5, высушенной с помощью распылительной сушки, приведена на фиг. 1. На рентгенограмме отсутствуют какие-либо отчетливые отражения,имеющие FWHM менее 2,5, что указывает на то, что композиция является рентгеноаморфной, в противоположность катализаторам согласно изобретению. На фиг. 2 и 3 приведены рентгенограммы триметаллического катализатора сравнительного примера С 4 и катализатора согласно изобретению примера Е 2. Сопоставление рентгенограмм на фиг. 1-3 четко демонстрирует описанные выше различия между аморфным NiMo катализатором прототипа, триметаллическим катализатором прототипа и катализатором согласно изобретению. Результаты испытания активности также показывают, что катализатор согласно изобретению имеет значительно более высокую активность при гидродеазотировании и более низкую концентрацию остаточного азота, чем аморфный катализатор сравнительного примера С 4. Сравнительный эксперимент С 6. Был воспроизведен способ, описанный в примере 6 в патентном документе WO 2004/073859. Растворяли 174 г Ni(NO3)26H2O (0,6 моль Ni) и 102,5 г ADM (0,6 моль Мо) в 1200 мл Н 2 О при комнатной температуре. При нагревании до 80 С добавляли 25,5 г HNO3. Получали прозрачный раствор с рН 2,56. Диспергировали 36,9 г Sipernat 2200 в 300 г Н 2 О и нагревали до 80 С. К раствору металла добавляли суспензию диоксида кремния. Медленно добавляли 7 мас.% NH4OH до рН 6,8, при котором происходило осаждение. Приблизительно через 30 мин после добавления диоксида кремния к раствору металла суспензию отфильтровывали. Осадок промывали. Вследствие потери Мо с фильтратом полученное твердое вещество имело состав 31,2 мас.% MoO3, 32,0 мас.% NiO и 36,8 мас.% SiO2. Полученное твердое вещество сушили при 120 С в течение 16 ч (в течение ночи). Полученный материал гранулировали, гранулы измельчали и отделяли просеиванием фракцию 40-60 меш. Материал затем прокаливали при 300 С в течение 1 ч. Материал затем сульфидировали и подвергали испытанию с использованием методики испытания дизельного топлива D4. На рентгенограмме высушенного твердого вещества отсутствуют какие-либо отчетливые отражения, что указывает на то, что композиция является полностью рентгеноаморфной. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Насыпной катализатор, включающий по меньшей мере 60 мас.% частиц оксидов металлов, содержащих один или более металлов группы VIII и молибден, включающий менее чем 10 мол.% любого другого металла группы VIB относительно суммарного количества металлов группы VIB и металл группы V в количестве менее чем 10 мол.% относительно суммы металлов группы VIB, где насыпной катализатор прокален при температуре ниже температуры, при которой гексагональная метастабильная кристаллическая структура превращается в неактивную кристаллическую структуру, и где насыпной катализатор имеет метастабильную гексагональную фазу, характеризуемую рентгенограммой, имеющей отражения при 33-35 и 58-612. 2. Насыпной катализатор по п.1, в котором молярное отношение металла группы VIII к металлу группы VIB составляет более 1,5. 3. Насыпной катализатор по п.1, в котором молярное отношение металла группы VIII к металлу группы VIB составляет от 2,5 до 5. 4. Насыпной катализатор по п.1, у которого главные отражения имеют полную ширину на половине максимума менее чем 2,5. 5. Насыпной катализатор по п.1, в котором металлом группы V является ниобий. 6. Насыпной катализатор по п.1, в котором частицы оксидов металлов практически включают только один металл группы VIII. 7. Способ получения насыпного катализатора по п.1, включающий:(i) приготовление реакционной смеси, состоящей из протонной жидкости, одного или более соединений первого металла, включающего один или более металл группы VIII, и соединения, включающего молибден, и меньше чем 10 мол.% любого другого металла группы VIB относительно суммарного количества металлов группы VIB и/или металла группы V в количестве меньше чем 10 мол.% относительно суммы металлов группы VIB,(ii) осуществление взаимодействия соединений первого и второго металлов при повышенной температуре, при котором твердые соединения первого и/или второго металла остаются, по меньшей мере,частично в твердом состоянии в процессе всей реакции с образованием частиц оксидов металлов,где насыпной катализатор на всех стадиях в ходе его приготовления остается при температуре ниже температуры, при которой гексагональная метастабильная кристаллическая структура превращается в неактивную кристаллическую структуру. 8. Способ по п.7, в котором соединение первого металла и соединение второго металла остается, по меньшей мере, частично в твердом состоянии в процессе всей реакции. 9. Способ по п.8, в котором соединением первого металла является гидроксикарбонатом металла и соединением второго металла является оксид металла или кислота. 10. Способ по п.9, в котором соединением первого металла является карбонат или гидроксикарбонат никеля, имеющий площадь поверхности по меньшей мере 150 м 2/г. 11. Способ получения насыпного катализатора по п.1, включающий:(i) приготовление раствора одного или более растворимых соединений первого металла, включающих один или более металлов группы VIII, и растворимого соединения второго металла, включающего молибден, и менее чем 10 мол.% относительно суммарного количества металлов группы VIB и/или ме- 14012365 талл группы V в количестве менее чем 10 мол.% относительно суммы металлов группы VIB,(ii) осуществление взаимодействия и/или осаждение соединений первого и второго металлов с образованием частиц оксидов,где насыпной катализатор на всех стадиях в процессе его приготовления остается при температуре ниже температуры, при которой гексагональная метастабильная кристаллическая структура превращается в неактивную кристаллическую структуру. 12. Способ по п.7, в котором протонной жидкостью является вода, и реакцию проводят при гидротермальных условиях при температуре реакции выше температуры кипения протонной жидкости. 13. Способ по п.7, в котором твердые соединения первого и второго металлов не содержат атомов азота и в котором протонную жидкость, отделенную от прореагировавших частиц оксидов металлов на стадии iii, повторно используют, по меньшей мере, частично для получения суспензии на стадии i. 14. Способ по п.7, дополнительно включающий одну или более из следующих стадий способа:(iii) отделения частиц оксидов металлов от реакционной смеси,(iv) необязательное смешение частиц оксидов металлов с от 0 до 40 мас.% одного или более материалов, выбранных из группы связующих материалов, традиционных катализаторов гидропереработки,кислотных промоторов и крекирующих соединений, до, во время или после смешения и/или взаимодействия соединений металлов,(v) распылительной сушки, (мгновенной) сушки, измельчения, замешивания, суспензионного смешения, сухого или мокрого смешения или их комбинации,(vi) формования,(vii) сушки и/или термической обработки при температуре ниже температуры, при которой происходит переход в кристаллическую структуру, предпочтительно ниже 450 С и(viii) сульфидирования. 15. Способ гидропереработки углеводородного сырья, включающего серосодержащие и азотсодержащие органические соединения, включающий стадию контактирования углеводородного сырья с катализатором по п.1.
МПК / Метки
МПК: B01J 23/883, B01J 37/03, B01J 23/88, B01J 23/887, C10G 45/08, B01J 35/00
Метки: насыпной, катализатор
Код ссылки
<a href="https://eas.patents.su/17-12365-nasypnojj-katalizator.html" rel="bookmark" title="База патентов Евразийского Союза">Насыпной катализатор</a>
Способ выщелачивания халькопирита в насыпной массе с использованием бактерий

Номер патента: 4713
Опубликовано: 24.06.2004
Автор: Хантер Колин Джон
МПК: C22B 3/18
Метки: насыпной, массе, бактерий, выщелачивания, халькопирита, использованием, способ
Формула / Реферат:
1. Способ выщелачивания халькопирита в насыпной массе с использованием бактерий, отличающийся тем, что сооружают насыпную массу руды, содержащую халькопирит, в которой осуществляют окисление содержащихся в ней сульфидных минералов, причем насыпная масса руды содержит и/или инокулирована сульфидокисляющей бактериальной культурой, которая либо не окисляет двухвалентное железо до трехвалентного, либо делает это неэффективно, используют по меньшей...
Способ выщелачивания руды в насыпной массе с помощью бактерий

Номер патента: 3499
Опубликовано: 26.06.2003
Автор: Хантер Колин Джон
МПК: C22B 3/18
Метки: бактерий, насыпной, способ, массе, руды, выщелачивания, помощью
Формула / Реферат:
1. Способ выщелачивания в насыпной массе с помощью бактерий, отличающийся тем, что сооружают насыпную массу руды для окисления содержащихся в ней сульфидных минералов, сооружают биологический контактор с посевом бактерий, окисляющих двухвалентное железо, сооружают, по меньшей мере, один бассейн для подачи выщелачивающего раствора к насыпной массе и биологическому контактору и приема выщелачивающего раствора из насыпной массы и биологического...
Способ получения композиций моющих средств с высокой насыпной плотностью

Номер патента: 1705
Опубликовано: 25.06.2001
Авторы: Эсер Хуг, Аппел Петер Виллем, Вагнер Хеннинг
МПК: C11D 17/06
Метки: моющих, получения, плотностью, высокой, средств, композиций, насыпной, способ
Формула / Реферат:
1. Способ получения гранулированной композиции моющего средства, включающий первую стадию получения жидкого компонента, содержащего структурирующую добавку в форме мыла, вторую стадию смешивания жидкого компонента с твердым компонентом в грануляторе, и, при необходимости, третью стадию сушки и/или охлаждения, при этом структурирующую добавку вводят в таком количестве, чтобы жидкий компонент обладал способностью к перекачиванию насосом при...
Катализатор для гидрирования ненасыщенных полимеров

Номер патента: 12191
Опубликовано: 28.08.2009
Автор: Ауезов Алий Байдильдаевич
МПК: B01J 21/06, B01J 21/08, B01J 21/10...
Метки: катализатор, ненасыщенных, гидрирования, полимеров
Формула / Реферат:
1. Катализатор для гидрирования ненасыщенных полимеров, содержащий палладий, нанесенный на оксиды магния, или лантана, или циркония, в сочетании с оксидом другого элемента, выбранного из группы, включающей кальций, магний, лантан, алюминий, титан, кремний, ванадий, хром и марганец, отличающийся тем, что он дополнительно включает молибден при следующем составе катализатора, мас.%: 2. Катализатор для гидрирования ненасыщенных полимеров по п.1,...
Катализатор

Номер патента: 9506
Опубликовано: 28.02.2008
Авторы: Лорен Этьенн, Сиро Даниель
МПК: C08F 255/02, C08F 297/08, C08F 10/02...
Метки: катализатор
Формула / Реферат:
1. Способ полимеризации олефинов в двух заполненных жидкостью реакторах с циркуляцией, соединенных последовательно, в котором производят фракции с различной молекулярной массой в присутствии каталитической системы Циглера-Натта, отличающийся тем, что катализатор Циглера-Натта имеет распределение частиц по размерам d50 менее 20 мкм и более 5 мкм. 2. Способ по п.1, в котором катализатор Циглера-Натта имеет d50 менее 15 мкм. 3. Способ по п.1 или 2,...
Предыдущий патент: Защитное устройство для защитных подложек
Следующий патент: Гидравлическое устройство
Случайный патент: Сплав на основе палладия 500 пробы